In the reaction shown in the equation below eighteen compounds are represented of which four (elemicin1, Ia; isoelemicin2, IIIa; asarone3, IIIb; and β-asarone4, IIb), as well as an undesignated "calamol"5, have been observed as constituents of essential oils, and another four have, in addition, been encountered synthetically as chemical intermediates6. The syntheses and physical properties of these and of the ten remaining isomers are described in this note. The methylation of the Claisen rearrangement product of allyl 2,6-dimethoxyphenyl ether yielded elemicin (Ia) which was identical with that isolated from nutmeg oil7 and the 2,4,6- counterpart (If) has been synthesized6a by methylation of the rearrangement product of allyl 3,5-dimethoxyphenyl ether. The synthesis of 2,3,4-trimethoxyallylbenzene (Ic) was achieved through the corresponding rearrangement of allyl 2,3-dimethoxyphenyl ether6b but it has been neither isolated nor characterized.
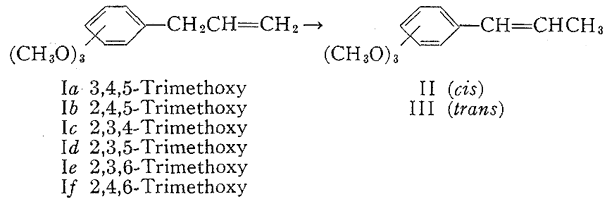
The three remaining isomers, Ib, Id, and Ie, were obtained by the thermal rearrangement of the appropriate allyl dimethoxyphenyl ethers, followed by methylation of the potassium salts of the resulting phenols with methyl iodide. In the first of these, the rearrangement of allyl 3,4-dimethoxyphenyl ether to the phenolic precursor of Ib, there are two ortho positions available. Rearrangement occurred preferentially (7:1) to the side nonadjacent to the methoxyl group, as shown by eventual conversion into asarone (IIIb) which was identical with that obtained from parsley oil In the latter two required rearrangements (those of allyl 2,4-dimethoxyphenyl ether and 2,5 counterpart to yield the phenolic precursors to Id and Ie respectively), only a single ortho position is available for the migrating allyl group. The first of these ethers, indeed, rearranged exclusively (>95%) to the available ortho position, but the second underwent appreciable para rearrangement (57% ortho, 43% para) as well as diallylation through some intermolecular phenomenon (see Experimental).
Twelve isomers (IIa-IIf ; IIIa- IIIf) can result from the base-catalyzed isomerization of the six trimethoxylallyl benzenes, Ia-If. IIb, IIIa and IIIb have been mentioned above as natural products, and both IIIc and IIIf have been prepared synthetically6. The hydroxide to mixtures of their cis- and trans-propenyl counterparts. The ratio of isomers that are formed invariably favored the trans- form, which further was found to be the slower moving component in gas-liquid chromatography (GLC) separations8 and to be, in all cases except one, a crystalline solid at or near room temperature. The isomers were purified by preparative GLC and only materials so obtained were utilized for the physical data recorded in Table 1.
Table I
Physical data of the Trimethoxypropenylbenzenes
| Methoxy position |
Alkyl group | Refractive indexa |
Melting pointb |
Retention timec |
Isomer ratio, trans/cisd |
| 3,4,5 | Allyl | 1.5308 |
17-18°C |
2.38 min |
11.4 |
| trans-Propenyl | 1.5487 |
2.78 min |
|||
| cis-Propenyl | 1.5572 |
3.94 min |
|||
| 2,4,5 | Allyl | 1.5317 |
57-58°C |
2.59 min |
11.5 |
| trans-Propenyl | 1.5559 |
3.27 min |
|||
| cis-Propenyl | 1.5652 |
4.70 min |
|||
| 2,3,4 | Allyl | 1.5174 |
25-26°C |
1.37 min |
5.4 |
| trans-Propenyl | 1.5345 |
1.69 min |
|||
| cis-Propenyl | 1.5460 |
2.28 min |
|||
| 2,3,5 | Allyl | 1.5240 |
44-45°C |
2.24 min |
13.4 |
| trans-Propenyl | 1.5394 |
2.53 min |
|||
| cis-Propenyl | 1.5495 |
3.68 min |
|||
| 2,3,6 | Allyl | 1.5238 |
|
1.73e min |
12.2 |
| trans-Propenyl | 1.5340 |
1.78e min |
|||
| cis-Propenyl | 1.5529 |
2.81 min |
|||
| 2,4,6 | Allyl | 1.5306 |
72-73°C |
2.62 min |
5.8 |
| trans-Propenyl | 1.5342 |
3.86 min |
|||
| cis-Propenyl | 1.5638 |
5.32 min |
Notes:
(a) All values measured with the sodium D line at 24.5°C.
Solids were supercooled.
(b) Uncorrected.
(c) Determined on a 20M polyethylene glycol column, described
in the Experimental. 1,2,3-Trimethoxybenzene was added to the test mixtures,
and its retention time taken as 1.00.
(d) Determined from the areas of the appropriate peaks,
as measured by a rolling-disk planimeter.
(e) Complete separation is obtained when a SE-30 silicone
substrate is employed.
Experimental:
The GLC separations were achieved on a Wilkens Instrument Company Autoprep A-700. The principle column employed was an aluminum column (5' x 3/4" diameter, loaded with 10% 20M polyethylene glycol on 60/80 mesh Chromosorb W. ) At an air bath temperature of about 180°C and with He flow rate of about 500ml/min, the standard internal reference chemical (1,2,3-trimethoxybenzene) required about 4 min to emerge. The retention times of materials summarized in this report may thus be estimated from the values in Table1. Other substrates employed in the work will be described as they are mentioned. Samples for microanalytical evaluation were obtained from the GLC separation directly in sealable capillaries. The members of this series all carry the empirical formula C12H15O3 and all analyses were acceptable.
All infrared spectra described in this report were synthesized by the general sequence of reactions; dimethoxyphenol → allyl dimethoxyphenyl ether → allyl dimethoxyphenol → trimethoxyallylbenzene (I) → trimethoxypropenylbenzene (II and III). As it would be unnecessarily repetitious to describe all six parallel processes, only one will be presented in detail. Any significant deviations required in the other synthesis will be mentioned briefly:
2,4-Dimethoxyphenol
A solution of 68g of 2,4-dimethoxybenzaldehyde (K&K Labs) in 250g of glacial acetic acid was brought to 25°C and treated dropwise, over 1 hour, with 86g of a 40% solution of peracetic acid in acetic acid. The reaction mixture was diluted with three volumes of water, and then neutralized cautiously with 283g of anhydrous potassium carbonate. Extensive extraction with ether yielded, after removal of the solvent, 66g of the formate of 2,4-dimethoxyphenol.
This was hydrolyzed with 10% base (Note: No base specifically mentioned, probably NaOH) for 1 hour on a steam bath. The resulting solution was extracted once with DCM (discarded), acidified with aq. HCl and extracted several times with DCM. The extracts were washed once with sodium bicarbonate solution (this removed most of the color), dried, and evaporated on a steam bath. The resulting product, which crystallized when cooled below RT, weighed 34.4g (54%) and yielded a benzoate (benzoyl peroxide in pyridine- recryst. from cyclohexane) with mp 89-90°C. (lit. values are : phenol, mp 28°C; benzoate, mp 90°C9). The 2,3-; 3,4-; and 2,5-dimethoxybenzaldehydes were oxidized similarly to the phenols, in similar yields. 3,5-dimethoxybenzaldehyde gave no phenolic product with peracetic acid, and the corresponding phenol was synthesized from phloroglucinol10 2,6-dimethoxyphenol was obtained commercially from the Eastman-Kodak Company.
Allyl-2,4-dimethoxyphenyl Ether
A solution of 31.0g of the above crude phenol in 60ml of absolute ethanol was combined with another containing 11.25g of potassium hydroxide in 90ml of anhydrous ethanol. Allyl bromide was immediately added (28g) and the resulting solution refluxed on the steam bath for 2 hours. A precipitate of potassium bromide formed within a minute of heating. At the end of the reaction, the mixture was quenched with several volumes of water, made strongly basic with 10% caustic, and extracted with several volumes of ether. Evaporation of the solvent provided 33.2g of the desired product (85% yield, bp 107-110°C/1.0mmHg; nD22.8 = 1.5322), which was shown to be free of the starting phenol by analysis on an ethylene glycol succinate GLC column (5% substrate, 10' x 3/8” diameter, 165°C (It has been found that the thermally unstable allyl ethers can be analyzed if chromatographed on an ethylene glycol succinate column at temperatures below 180°C. Under these conditions there is no detectable rearrangement to the allyl phenols. With a silicone column, even at these temperatures, there is extensive continuous rearrangement on the column, providing a complex and misleading picture of the original composition.)
2-Allyl-4,6-dimethoxyphenol
The ether (31.0g) was heated 215°C, at which temperature an exothermic reaction ensued that raised the temperature to 270°C. The temperature was held at about 260°C for an additional 10minutes. The rearrangement product thus obtained (nD25 = 1.5464) was submitted to methylation without further purification (The presence of about 10% of a 2,4-dimethoxyphenol in this product (a material specifically shown to be absent in the ether precursor) indicates a side reaction involving deallylation. The phenol is best removed as its methyl ether (1,2,4-trimethoxybenzene) in the next step.)
2,3,5-trimethoxyallylbenzene
The phenol was methlated directly, in anhydrous ethanol, by treatment with first, 8.7g of potassium hydroxide dissolved in 75ml of boiling ethanol, and then with 22.4g methyl iodide. Potassium iodide formed immediately. After 3 hours of reflux, the reaction mixture was cooled, quenched with several volumes of water made strongly basic with 10% caustic, and extracted with 4 x 100ml ether. These extracts, after drying and removal of the solvent by evaporation, provided 28g of an oil. Analysis by GLC showed the presence of about 10% 1,2,4-trimethoxybenzene (from the 2,4-dimethoxyphenol known to be present before methylation) and less than 2% all other volatile contaminants. The yield of Id was thus greater than 80% based on the initial allyl ether. The infrared spectra of the crude reaction product and of the chromatographically isolated specimen were virtually identical; the latter was employed for the refractive index value.
The 2,3,6-trimethoxy isomer obtained similarly from allyl-2,5-dimethoxyphenyl ether was admixed with an almost equal amount of the 2,4,5-isomer. To obtain an adequate amount of the former material both for necessary descriptions and for the subsequent isomerization to the propenyl mixture, it was necessary to repeatedly inject scores of 0.15ml portions into the Wilkins machine, utilizing the existing trapping machinery. Trapping efficiency was less than 50% because of extensive aerosol formation. In this process, a later peak, constituting 12% of the entire crude reaction mixture, was trapped. This trimethoxydiallylbenzene showed a nD24.5 = 1.5228).
2,3,5-trimethoxypropenylbenzene
A solution of 26g of the crude trimethoxyallylbenzene in an equal weight of anhydrous ethanol was treated with 52g of finely flaked potassium hydroxide. Heating on the steam bath for 24 hours with occasional agitation effected complete isomerization to the propenyl mixture. The reaction mixture was quenched with much water, and extracted with ether. Removal of the solvent provided 24.6g of the crude mixture with a yield of about 95%. Dilution with pentane, intense cooling and rapid filtration gave 9.2g of an amber-colored crystalline solid. This, upon recrystallization from hexane, provided the pure trans isomer, mp 44-45°C. The mother liquors of the pentane crystallization were stripped of solvent to give the cis isomer. This was subjected to GLC. The 2,3,4- and 2,3,6- trimethoxyallylbenzenes required twice as long for the isomerization.