Ephedrine was discovered in 1887, but it attracted attention only after it came into use in medicine as a sympathomimetic agent. Initially it was obtained exclusively from the Chinese plant Ma Huang, but later it was discovered in several varieties of Ephedra1. The total alkaloid contents of various Ephedra species containing ephedrine vary from 0.2 to 2.5%2. In the USSR (Kazakhstan, Kirghizia, and other republics) the total alkaloid contents of various Ephedra species are 1.5-2%3.
Among the various syntheses of the ephedrine alkaloids, the following are worthy of mention. Sp?th and G?hring4 synthesized both racemic forms of ephedrine from propionic aldehyde, bromine, and phenylmagnesium bromide:
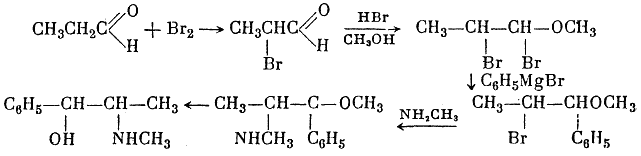
They obtained DL-pseudoephedrine, which was isomerized to ephedrine. Ebergard5 started with propiophenone; the α-methylaminopropiophenone formed was reduced by sodium amalgam in presence of dilute hydrochloric acid and by hydrogen in presence of palladium on wood charcoal. The main product was pseudoephedrine. In 1929 Fourneau6 condensed benzene with the acid bromide of α-bromopropionic acid. The resultant α-bromopropiophenone was converted into the secondary amine, which yielded ephedrine on reduction with hydrogen (over platinum black catalyst). Somewhat later two groups7,8, working independently, subjected an equimolar mixture of α-phenyl-α,β-dioxypropane and methylamine in alcoholic solution to catalytic hydrogenation by the action of hydrogen in the presence of colloidal platinum:
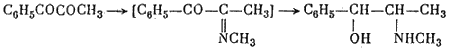
The reaction yields DL-ephedrine with a small admixture of DL-pseudo- ephedrine. A disadvantage of this method, noted by Temnikova9 who verified it, is the cumbersome preparation of the starting material.
We used propionic acid as the starting material for synthesis of ephedrine, as this acid is a by-product in the manufacture of acetoacetic ester and can be easily obtained from acrylonitrile, which is produced in our country on a large scale.
The reaction scheme given below shows that propionic acid is chlorinate by phosphorus trichloride; phosphorous acid is separated from the resultant solution of propionyl chloride in benzene, and the benzene solution reacts further with aluminum chloride to give propiophenone in 83-85% yield, calculated on the propionic acid taken (a).
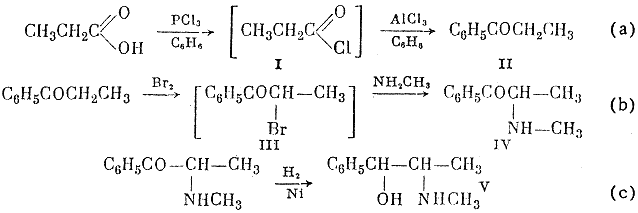
From the aqueous alkaline solution after amination 11 g of a mixture of methylamine hydrochloride and hydroromide was isolated.
The α-bromopropiophenone (III) formed by reaction (b) is methylaminated in the same vessel.; this reaction gives α-methylaminoethyl phenyl ketone (ephedrone) in 70-74% yield on the propiophenone taken. Ephedrone is reduced by molecular hydrogen in presence of Raney nickel catalyst (c). The yield of DL-ephedrine in this reaction, calculated on the ephedrone taken, is 64%; 19% of DL-pseudophedrine, to be converted into DL-ephedrine, is also obtained.
Experimental
Preparation of Propionyl Chloride (I) and Propiophenone (II)
A round flask fitted with a stirrer and reflux condenser contained 37 g (0.5 mole) of propionic acid, 40 ml of dry benzene, and 29 g (0.21 mole) of phosphorus trichloride; the reaction mass was stirred at 55-62?C for 1 hour. The mixture was then cooled to 30?C and phosphorous acid was separated off. To the remaining benzene solution 25 ml of dry benzene was added, the mixture was shaken, and the residual phosphorous acid was separated off.
Dry benzene (85 g) and 65 g of anhydrous aluminum chloride were put in a flask fitted with a stirrer, a reflux condenser, a dropping funnel, and a thermometer. The previously prepared solution of propionyl chloride in benzene was added to this mixture gradually during 3 hours at 35-45?C. The reaction mixture was heated at 45-50?C for 2 more hours, cooled to 30?C, and poured into a mixture of 300 g of ice and 50 g of concentrated hydrochloric acid. After the mixture had settled out the solution of propiophenone in benzene was separated off, made slightly alkaline to litmus by addition of 20% sodium carbonate solution, and distilled with steam. The solution of propiophenone in benzene was separated from the distillate and dried over calcium chloride; the benzene was then distilled off and the residual propiophenone was distilled under vacuum at 10 mm and 103-105?C. The yield of propiophenone was 56.5 g (84% of the propionic acid taken).
Ephedrone [α-Methylaminoethyl Phenyl Ketone (IV)]
A flask fitted with a stirrer, a dropping funnel, a thermometer, and an air inlet tube contained 56.5 g (0.421 mole) of propiophenone and 130 ml of benzene; 67.5 g (0.421 mole) of bromine was added to the mixture at 30?C at such a rate that the bromine became decolorized. If the decolorization of the reaction mixture was slow, air was blown through it to remove the hydrogen bromide formed. After the bromine had been added the solution was stirred for 30 minutes and hydrogen bromide was then blown out of the solution with the stirrer running. After removal of hydrogen bromide 30 ml of water was added to the solution, the mixture was stirred thoroughly, and 20% sodium carbonate solution was added until the mixture was weakly alkaline to litmus. A solution of 56.5 g (0.835 mole) of methylamine hydrochloride in 55 ml of water was then added. A solution of 67.5 g (1.67 moles) of caustic soda in 80 ml of water was added during 10 minutes at 50-60?C; the temperature was raised to 70?C. At the end of the reaction the mixture was cooled to room temperature; unconverted methylamine was blown out by air into absorption flasks containing 5% hydrochloric acid. The benzene and aqueous alkaline layers were then separated, the latter was extracted twice with benzene, and the extracts were added to the main solution. This solution was washed with water and stirred with 1 N hydrochloric acid solution. The resultant aqueous solution of ephedrone hydrochloride was evaporated under vacuum to a thick syrup; this was stirred with acetone to yield a white precipitate of ephedrone hydrochloride. The precipitate was heated to boiling with acetone and cooled; the white crystals were filtered off, washed with acetone, and dried.
A certain amount of less pure ephedrone hydrochloride was additionally obtained from the mother liquors and crystallized from a mixture of ethyl alcohol and acetone. The total amount of ephedrone hydrochloride, mp 175-179?C, was 59-62 g (70-74% on the propiophenone taken).
The solution of methylamine hydrochloride collected in the absorption flask was evaporated to dryness; 21 g of methylamine hydrochloride was recovered, mp 220-222?C (literature value, 225-226?C10).
DL -Ephedrine Hydrochloride (V)
A solution of 14.2 g of caustic soda in 135 ml of ethyl alcohol was added to a solution of 62 g of ephedrone hydrochloride to give an alkaline reaction to phenolphthalein; the precipitated sodium chloride was filtered off and washed with alcohol; the washings were added to the main alcoholic solution of ephedrine base. This solution was reduced by molecular hydrogen in presence of Raney nickel catalyst at room temperature and atmospheric pressure. At the end of the reduction the solution was cooled to 5-10?C the catalyst was filtered off, and the solution was acidified with a solution of hydrogen chloride in alcohol; the precipitate sodium chloride was filtered off and the filtrate was evaporated under vacuum at a temperature not higher than 45?C The yield was 40 g of DL-ephedrine hydrochloride, mp 184-187?C After crystallization from ethyl alcohol the crystals had mp 187-190?C (literature value 187?C5).
After treatment of the mother liquors the total yield of DL-ephedrine hydrochloride was 64% on the ephedrone taken. In addition, 19% of pseudoephedrine was obtained.