
The by far easiest method to synthesize tryptamine is the decarboxylation of the amino acid tryptophan. Both the natural form of tryptophan (L-tryptophan) as well as the synthetic form (DL-tryptophan) can be used with the same good results, both variants are fully interchangeable in the syntheses below.
Purification of Tryptamine
A good way to purify tryptamine without having to resort to distillation under strong vacuum is to dissolve the crude tryptamine hydrochloride in water, adjust the pH to between 7.6 and 8.2 and extract the solution with chloroform. The pH is then adjusted to 14 with NaOH and the pure tryptamine is filtered off with suction and air dried.
Experimental
Decarboxylation in Diphenyl Ether1
DL-Tryptophan (1.0 g) and diphenyl ether (50 ml) were heated at reflux for 1 hour in an atmosphere of nitrogen. The mixture was cooled and extracted with 2N aqueous hydrochloric acid (3x40ml). This extract was washed with ether, basified (6N NaOH), and extracted with ether (5x50ml). This extract was washed with water and brine, dried over sodium sulfate, and the solvent removed in vacuo, leaving a residue which was recrystallized from benzene to give pale yellow prisms (530mg), mp 113-114°C. Sublimation afforded a colorless crystalline solid (450mg, 57%), mp 114-115°C.
The use of freshly distilled tetralin as the solvent for decarboxylation led to a yield of only 36%. With commercial tetralin the yield was reduced to 20%. No tryptamine was isolated from experiments which employed diphenylamine or dimethylsulfoxide in place of diphenyl ether.
Decarboxylation of Tryptophan in Diphenylmethane2
A suspension of L-tryptophan (250 mg) in warm diphenylmethane (10 g) was gently refluxed in a stream of nitrogen for 5-20 min until no more evolution of carbon dioxide was observed. After cooling, the clear pale yellow reaction mixture was treated with a benzene solution (20 ml) saturated with dry hydrogen chloride. The resulting precipitate was collected by filtration, washed with n-hexane and dried to afford crude tryptamine hydrochloride (223 mg, 93%) which was recrystallised from ethanol/ethyl acetate to yield tryptamine hydrochloride (151 mg, 63%) as colorless needles, mp 248-249°C.
Another similar procedure (unfortunately without reference), reads
as follows:
A mixture of 0.3-0.5 g of DL-tryptophan and 12-20 g
of diphenylmethane was boiled over a burner flame in an atmosphere of nitrogen
for 20 min. After cooling, 20-40 ml of a saturated benzene solution of hydrogen
chloride was added to the mixture. The precipitate of salts that deposited
was separated off and was dissolved in a mixture of ethanol and ethyl acetate.
On strong cooling, lustrous colorless crystals deposited with a mp 248-249°C.
The experiment was repeated several times. Yield 75-90%.
Copper-catalyzed Decarboxylation of Tryptophan3
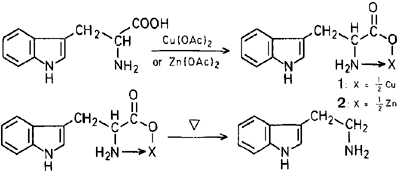
Tryptophan Copper Chelate
To a solution of L-tryptophan (50g) in water was added a solution of an excess of copper(II)acetate in water. The resultant precipitate was filtered. The extract was then washed several times with hot water to give the copper chelate compound. Yield: 52g, mp >280°C.
Decarboxylation of the Tryptophan Copper Chelate
A suspension of Tryptophan Copper Chelate in DMSO was heated at 170-175°C for several minutes, during which time an evolution of carbon dioxide was observed. After cooling, the resultant precipitate was filtered and to the filtrate was added a suitable amount of water. The reaction mixture was made basic with 30% sodium hydroxide solution and extracted with chloroform. After distillation of the solvent, the resultant residue was purified by flash chromatography on silica gel to givce tryptamine in 40% yield. The use of HMPA (hexamethylphosphoric triamide) instead of DMSO increased the yield to 45%, but that small increase in yield is not worth working with the expensive and highly toxic solvent HMPA.
Decarboxylation of Tryptophan in Tetralin With a Ketone Catalyst4
L- or DL-Tryptophan (102.1 g, 0.5 mol) was suspended in tetralin (250 ml) containing acetone (2.9g, 0.5 moles) and the mixture was heated to reflux for 8-10 hours with vigorous stirring until no more carbon dioxide was evolved and the reaction mixture became clear. The solvent was removed under vacuum, and the residue was distilled under reduced pressure to give a yellow crystalline solid, bp 140-155°C at 0.25 mmHg. This was recrystallized from boiling benzene to afford faint yellow prisms, mp 116-117.5°C (lit 115-117°C).
The yield with acetone as catalyst was 75%, methyl ethyl ketone 84.4%, 3-pentanone 85% and 2-pentanone 86.2%.
Ketone-catalyzed decarboxylation, as described by Drone #342:
Decarboxylation is accomplished by mixing about 80 g tryptophan in 250 mL
of high-boiling solvent (xylene, DMSO, cyclohexanol, etc.), adding a dash
of a ketone (I like 5 g of cyclohexanone, but a couple grams of MEK works
reasonably well), heat it to around 150 deg, and when evolution of CO2
ceases/solution is clear, the reaction is complete. This takes anywhere
from 1.5 to 4 hours. After this is over, the solvent is boiled off (or at
least greatly reduced in volume), and the residue is dissolved in DCM. This
is washed with a 5% NaHCO3 solution, then a distilled water solution,
then the DCM layer is separated off, dried with MgSO4, and the
DCM is boiled off. You now have reasonably pure tryptamine.
Decarboxylation in Cyclohexanol, with 2-Cyclohexen-1-one as catalyst5
20 grams of L-Tryptophan was dissolved in 150 ml cyclohexanol containing 1.5 ml of 2-cyclohexen-1-one, and the temp of the solution was held at 154°C for 1.5 hours. The tryptamine was isolated as the HCl salt, mp 256°C. Yield 92.3%.
Spearmint Oil Catalyzed Decarboxylation of Tryptophan
by "Student"
A mixture of 75 mL of turpentine (1), 7.14 grams of L-tryptophan (2), and 15 drops (0.25 grams; 0.3 mL) of spearmint oil (3) were placed in a 250 mL Erlenmeyer flask. A water cooled reflux condenser (4) was attached to the flask by a rubber stopper (5). The mixture in the flask was boiled (6)fast enough that there was at least one drop returning to the flask from the condenser every second. The mixture became transparent in four hours and heating was turned off after another 30 minutes. There was a little yellow solid on the side of the flask above the liquid. After sitting overnight there was a clump of yellow crystals in the corner of the flask and solidified dark oil across the bottom. The flask was refrigerated for the day and the orangish mother liquor was poured off.
The impure tryptamine was purified as follows (7). To the flask were added 150 mL of 5% distilled household vinegar along with 5 mL of chloroform (8) and the flask was briskly swirled until all solid was gone and there was only a little dark brown oil not dissolved in the yellow suspension. The hazy yellow liquid (pH 5-6) upper layer was filtered through a plug of cotton. The small amount of dark brown lower organic layer was extracted with another 10 mL of vinegar, and the resulting upper layer was filtered through the cotton plug. To the combined filtrates were added 5 mL of chloroform and enough sodium bicarbonate (10.58 g) in portions so that further addition caused very little foaming. The flask was swirled thoroughly and the hazy yellow aqueous upper layer was filtered through a fresh plug of cotton. The filtrate was cooled in the freezer for 15 minutes, basified with 12 mL of 25% sodium hydroxide solution, and set back in the freezer for 30 minutes. The solid was dislodged from the sides with a metal scoop and the mixture was filtered through filter paper (9). The flask and crystals were rinsed with 100 mL of ice cold household ammonia in portions (10). The filter paper was pressed between paper towels until damp and set aside to dry. The light yellow crystals weighed 3.64 grams (65% yield).
The turpentine mother liquor from the last reaction, still containing spearmint oil and some tryptamine, was used directly to decarboxylate 7.23 grams of L-tryptophan. This time the reaction took seven hours to become transparent, so apparently some of the catalyst was consumed during the first reaction. This time both the turpentine and the solid product were extracted with vinegar as above, and brought through the same purification process, to give 5.21 grams (92% yield) of light yellow crystals. The combined yield of tryptamine for the last two reactions is 79%. The solid melted at 117-118.5°C (Merck 118°C) and had one tan spot (Rf ~0.1 - 0.2) on silica TLC, eluting with methanol containing ~50 mg of ammonium carbonate.
Notes:
- If xylene (bp 118°C) is used as the solvent, the reaction will require about one week to complete, but the product will probably separate as crystals and be purer than if turpentine is used. If turpentine (bp 154-170°C) is used, the reaction will take several hours, but part of the product may come out as an oil. If difficulty is encountered obtaining a solid from a reaction using turpentine, place the flask in boiling water to cool. After a few minutes add a seed crystal and let the water cool slowly with the flask in it. This may allow complete crystallization of the product.
- It is possible that 5-hydroxytryptophan (5-HTP) would successfully decarboxylate under these conditions, giving 5-hydroxytryptamine (serotonin). However, obtaining pure starting material from the mixtures marketed as 5-HTP may be a challenge.
- The key to this reaction is the catalytic activity of carvone, an enone found in spearmint oil. The catalytic activity of enones was first reported by M. Hashimoto5. The spearmint oil (50% L-carvone) must be the pure essential oil, not an extract. Other oils which may also work are caraway (58% D-carvone), dill (50% D-carvone) or pennyroyal oil (85% pugelone). Successful decarboxylation using butanone under the above conditions (reported by another author) could not be reproduced. Good yields were reported in a Russian journal by refluxing tryptophan in acetophenone.
- A cork may be used instead of a rubber stopper. Cork doesn't seal as well as rubber, but it is less likely to contaminate and color the product.
- The rate of boiling needs to be sufficient to prevent the reaction mixture from being exposed to air. Air exposure causes a dark deposit to form on the flask walls, and no product can be isolated from it even by acid extraction. In a sufficiently spacious flask, the wall of the flask which is above the liquid surface can serve as an air condenser so that no condenser needs to be attached. Alternatively, an air condenser (air-cooled glass tube) is very effective with high boiling liquids, and even xylene boils high enough in this case to be made to work. The important thing is to keep the vapor part of the way up the tube, so that air doesn't reach the reaction mixture nor does vapor climb to the mouth of the tube (heating the entire tube to xylene's bp) and escape. Using the solvent vapor to exclude air renders an inert atmosphere unnecessary.
- Magnetic stirring isn't required for this reaction if the boiling is sufficiently vigorous to keep the solid tryptophan suspended in the solvent. Boiling stones may also be unnecessary, since the tiny bubbles of carbon dioxide produced during decarboxylation serve as nuclei the way the bubbles in boiling stones do. Heat was provided by an electric hot plate with a sparkless electronic thermostat. Since these solvents are flammable, the use of flame heating is not recommended.
- This purification process was modeled after the tryptamine purification outlined in the Tryptophan and Tryptamine FAQ.
- There is what sounds like an excellent procedure for the preparation of chloroform from bleach and acetone in this document. Others have reported excellent results with trichloroethylene instead.
- Coffee filter paper will probably work instead of laboratory filter paper.
- The use of cold household (clear) ammonia for washing is very important. Washing with water will cause the product to quickly dissolve! ChemFinder.com lists the solubility of tryptamine in water as 34 g/L.
L-Tryptophan From Milk
Casein6
To 1 liter of milk, from which the cream has been largely separated (by simple skimming), 0.05 M hydrochloric acid is slowly added, with stirring through a capillary tube extending to the bottom of the beaker. The addition is continued until the solution attains a pH of 4.6 (casein exists in milk in the form of a calcium derivative; pH 4.6 is the isoelectric point of free casein, which is soluble to the extent of only 0.11g/L water). Approximately 1 L of acid is required; the separation of the casein is practically complete at this point. Three liters of water is then added, stirring is discontinued, and the flocculent precipitate of casein is allowed to settle in the refrigerator for twelve to twenty-four hours. The clear supernatant liquid which contains soluble proteins and salts is removed as completely as possible by siphoning; the precipitate is collected on a suction funnel and washed with cold distilled water until the washings are free of calcium (test with ammonium oxalate)
The casein, which is contaminated with calcium phosphate and fats; is filtered to as small a volume as possible (about 500 mL) and transferred to a 2000ml beaker. It is then treated with 0.1 M sodium hydroxide, the alkali being added slowly and with stirring through a capillary extending to the bottom of the beaker (it is important to avoid a local excess of alkali, which would tend to denaturate the casein). The addition of alkali is continued until the pH of the mixture reaches 6.3 (at this pH sodium caseinate is largely dissolved, whereas calcium caseinate is largely undissolved); 100-150 mL of the alkali is required. At this pH the casein is completely in solution in the form of its sodium salt; fats, calcium phosphate, and any calcium caseinate remain undissolved. Care must be taken not to add more alkali than is necessary to bring the pH to the above point. The milky solution is filtered through a thick layer (10-15 mm.) of filter paper pulp tightly packed upon a suction funnel. The filtrate may be slightly opalescent; if it is less clear it is again filtered through a fresh layer of pulp.
The filtrate is brought to a pH of 4.6 with 0.05 M hydrochloric acid just as in the original precipitation, the necessary amount of acid being determined by titration of an aliquot portion, diluted fivefold, with 0.01 M hydrochloric acid, 220-250 mL of 0.05 M acid is required. As the reprecipitation progresses, the rate at which the acid is added is decreased in order to prevent precipitation at the tip of the capillary tube; vigorous mechanical stirring is, of course, essential. When the acidification is complete, 5000ml of cold distilled water is added and the flocculent precipitate allowed to settle in the refrigerator. After siphoning off the clear supernatant liquid, the casein is collected on a suction funnel, using hardened paper, washed with cold distilled water until free of chloride, sucked as dry as possible, and dried over calcium chloride in a vacuum desiccator. The yield is 23-29 g. of a colorless coherent product which may readily be pulverized in a mortar.

L-Tryptophan7
In an 8 Liter bottle is placed 600g of commercial casein (coarse powder), which is then covered with about 3200 mL of tap water at 37°C. The bottle is shaken until all the casein is moistened. A solution of 60 g. of anhydrous sodium carbonate and 6 g. of sodium fluoride (to inhibit oxidase enzymes present) in 1 L of water at 37°C is added. A thin paste of 20 g. of commercial pancreatin in 100 mL of water (37°C) is poured in. The mixture is covered with a layer of toluene (80 mL), diluted to 6 L, stoppered, shaken thoroughly, and placed in a warm room or bath at 37°C. After four or five days, with daily shakings, most of the casein is in solution and chalky masses of tyrosine begin to separate. After five days, a second 20-g. portion of pancreatin in 100 mL of water is added. After twelve days, the bottle is cooled in an icebox overnight and the undissolved material is filtered off (This filtration may be slow. Büchner funnels of 20-cm. diameter are best used; the material from a single filling is allowed to suck dry and the filter paper then changed).
The filtrate (6.9-7 L) is measured into a 16-L stone jar, and for every liter there is added 163 mL of dilute sulfuric acid (one volume of 95 per cent sulfuric acid and one volume of water, cooled to room temperature). The first part of the acid must be added cautiously on account of the liberation of carbon dioxide. The tryptophan is precipitated by adding a solution of 200 g. of mercuric sulfate (Note 5) in a mixture of 1860 mL of water and 140 mL of 95 per cent sulfuric acid. After standing for twenty-four to fortyeight hours, the clear liquid is siphoned out and the yellow precipitate is filtered and washed with a solution of 100 mL of concentrated sulfuric acid in 1.9 L of distilled water containing 20 g. of mercuric sulfate, until the filtrate is colorless and Millon's test is atypical; about 1.5 L is necessary. The precipitate is washed with three successive 500-mL portions of distilled water to remove most of the sulfuric acid.
The moist precipitate (120-130 g) is suspended with mechanical stirring in 1.2-1.3 L of distilled water, and a hot, 20 per cent aqueous solution of barium hydroxide is added until the mixture is permanently alkaline to phenolphthalein (about 120 mL is required). A rapid stream of hydrogen sulfide is passed in with stirring until the mercury is completely precipitated. The precipitate is filtered and washed with water until a sample of the washings gives a negative test for tryptophan with bromine water. The barium is removed from the combined filtrate and washings by adding the exact amount of dilute sulfuric acid and filtering. The filtrate is concentrated under reduced pressure to about 80 mL.
The tryptophan is extracted from the aqueous solution by repeated shaking in a separatory funnel with 25-mL quantities of n-butyl alcohol; water is added from time to time to keep the volume approximately constant. The butyl alcohol extract is distilled under reduced pressure. After the water present has distilled, the tryptophan precipitates in the distilling flask and may cause bumping. When all the water has been removed, as is indicated by non-formation of drops on the side of the condenser, the distillation is stopped and, after cooling, the tryptophan is filtered and washed with a little fresh butyl alcohol. Such extractions and distillations are continued until the quantities of tryptophan obtained are negligibly small.
The tryptophan so produced (7-8 g.) varies somewhat in quality in different runs. It is purified by recrystallization from 60 mL of dilute alcohol (two volumes of 95% alcohol to one volume of water), filtering from the hot solution an appreciable quantity of insoluble matter, and subjecting this to a second extraction with an additional 10 mL of aqueous alcohol. The solution is decolorized by the addition of 1 g. of Norite and allowed to stand in the icebox; the silvery leaflets of tryptophan are filtered and washed successively with cold 70 per cent, 80 per cent, 95% alcohol, and, finally, with a little ether. Less than half the tryptophan is obtained in each crystallization. The yield of pure tryptophan is 4.0-4.1 g., together with under 0.1 g of less pure product.