

#146 3-TASB
3-THIOASYMBESCALINE; 4-ETHOXY-3-ETHYLTHIO-5-METHOXYPHENETHYLAMINE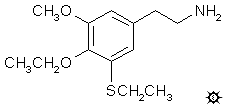
|
| [3D .mol structure] |
A solution of 32.5 g 3-bromo-N-cyclohexyl-4-hydroxy-5-methoxybenzylidenimine in 60 mL of hot DMF was cooled to near room temperature, treated with 24.5 g ethyl iodide and followed by 14.0 g of flake KOH. This mixture was held at reflux for 1 h, cooled, and added to 1 L H2O. Additional base was added and the product was extracted with 3x150 mL CH2Cl2. These pooled extracts were washed with dilute NaOH, then with H2O, and finally the solvent was removed under vacuum. The crude amber-colored residue was distilled. The fraction coming over at 118-135 °C at 0.4 mm/Hg weighed 8.7 g, spontaneously crystallized, and proved to be 3-bromo-4-ethoxy-5-methoxybenzaldehyde, melting at 59-60 °C after recrystallization from MeOH. Anal. (C10H11BrO3) C,H. The fraction that came over at 135-155 °C at 0.2 mm/Hg weighed 10.5 g and also solidified in the receiver. This product was 3-bromo-N-cyclohexyl-4-ethoxy-5-methoxybenzylidenimine which, upon recrystallization from two volumes MeOH, was a white crystalline material with a mp of 60-61 °C. Anal. (C16H22BrNO2) C,H. The two materials have identical mps, but can be easily distinguished by their infra-red spectra. The aldehyde has a carbonyl stretch at 1692 cm-1, and the Schiff base a C=N stretch at 1641 cm-1.
A solution of 20.5 g 3-bromo-N-cyclohexyl-4-ethoxy-5-methoxybenzylidenimine in about 300 mL anhydrous Et2O was placed in a He atmosphere, well stirred, and cooled in an external dry ice acetone bath to -80 °C. There was then added 50 mL of 1.6 N butyllithium in hexane. The mixture became yellow and very viscous with the generation of solids. These loosened up with continuing stirring. This was followed by the addition of 10.7 g diethyldisulfide. The reaction became extremely viscous again, and stirring was continued while the reaction was allowed to warm to room temperature. After an additional 0.5 h stirring, the reaction mixture was added to 800 mL of dilute HCl. The Et2O phase was separated and the solvent removed under vacuum. The residue was returned to the original aqueous phase, and the entire mixture heated on the steam bath for 2 h. The bright yellow color faded and there was the formation of a yellowish phase on the surface of the H2O. The aqueous solution was cooled to room temperature, extracted with 3x100 mL CH2Cl2, the extracts pooled, washed first with dilute HCl, then with saturated brine, and the solvent removed under vacuum. The residue was an amber oil weighing 20.4 g, and was distilled at 130-140 °C at 0.3 mm/Hg to yield 12.9 g of 4-ethoxy-3-ethylthio-5-methoxybenzaldehyde as a straw colored oil that did not crystallize. Anal. (C12H16O3S) C,H.
A solution of 1.0 g 4-ethoxy-3-ethylthio-5-methoxybenzaldehyde in 20 g nitromethane was treated with about 0.2 g of anhydrous ammonium acetate and heated on the steam bath. TLC analysis showed that the aldehyde was substantially gone within 20 min and that, in addition to the expected nitrostyrene, there were four scrudge products (see the discussion of scrudge in the extensions and commentary section under 3-TSB). Removal of the excess nitromethane under vacuum gave an orange oil which was diluted with 5 mL cold MeOH but which could not be induced to crystallize. A seed was obtained by using a preparative TLC plate (20x20 cm) and removing the fastest moving spot (development was with CH2Cl2). Placing this in the above MeOH solution of the crude nitrostyrene allowed crystallization to occur. After filtering and washing with MeOH, 0.20 g of fine yellow crystals were obtained which melted at 75-77 °C. Recrystallization from MeOH gave a bad recovery of yellow crystals of 4-ethoxy-3-ethylthio-5-methoxy-beta-nitrostyrene that now melted at 78.5-79 °C. Anal. (C13H17NO4S) C,H. This route was discarded in favor of the Wittig reaction described below.
A mixture of 27 g methyltriphenylphosphonium bromide in 150 mL anhydrous THF was placed under a He atmosphere, well stirred, and cooled to 0 °C with an external ice water bath. There was then slowly added 50 mL of 1.6 N butyllithium in hexane which resulted in the initial generation of solids that largely redissolved by the completion of the addition of the butyllithium and after allowing the mixture to return to room temperature. There was then added 11.7 g of 4-ethoxy-3-ethylthio-5-methoxybenzaldehyde without any solvent. There was the immediate formation of an unstirrable solid, which partially broke up into a gum that still wouldn't stir. This was moved about, as well as possible, with a glass rod, and then all was added to 400 mL H2O. The two phases were separated and the lower, aqueous, phase extracted with 2x75 mL of petroleum ether. The organic fractions were combined and the solvents removed under vacuum to give the crude 4-ethoxy-3-ethylthio-5-methoxystyrene as a pale yellow fluid liquid.
A solution of 10 mL of borane-methyl sulfide complex (10 M BH3 in methyl sulfide) in 75 mL THF was placed in a He atmosphere, cooled to 0 °C, treated with 21 mL of 2-methylbutene, and stirred for 1 h while returning to room temperature. This was added directly to the crude 4-ethoxy-3-ethylthio-5-methoxystyrene. The slightly exothermic reaction was allowed to stir for 1 h, and then the excess borane was destroyed with a few mL of MeOH (in the absence of air to avoid the formation of the dialkylboric acid). There was then added 19 g of elemental iodine followed, over the course of about 10 min, by a solution of 4 g NaOH in 50 mL hot MeOH. The color did not fade. Addition of another 4 mL 25% NaOH lightened the color a bit, but it remained pretty ugly. This was added to 500 mL H2O containing 5 g sodium thiosulfate and extracted with 3x100 mL petroleum ether. The extracts were pooled, and the solvent removed under vacuum to provide crude 1-(4-ethoxy-3-ethylthio-5-methoxyphenyl)-2-iodoethane as a residue.
To this crude 1-(4-ethoxy-3-ethylthio-5-methoxyphenyl)-2-iodoethane there was added a solution of 20 g potassium phthalimide in 150 mL anhydrous DMF, and all was held at reflux overnight. After adding to 500 mL of dilute NaOH, some 1.4 g of a white solid was generated and removed by filtration. The aqueous filtrate was extracted with 2x75 mL Et2O. These extracts were combined, washed with dilute HCl, and the solvent removed under vacuum providing 23.6 g of a terpene-smelling amber oil. This was stripped of all volatiles by heating to 170 °C at 0.4 mm/Hg providing 5.4 g of a sticky brown residue. This consisted largely of the desired phthalimide. The solids proved to be a purer form of 1-(4-ethoxy-3-ethylthio-5-methoxy)-2-phthalimidoethane and was recrystallized from a very small amount of MeOH to give fine white crystals with a mp of 107.5-108.5 °C. Anal. (C21H23NO4S) C,H. The white solids and the brown impure phthalimide were separately converted to the final product, 3-TASB.
A solution of 1.2 g of the crystalline 1-(4-ethoxy-3-ethylthio-5-methoxyphenyl)-2-phthalimidoethane in 40 mL of warm n-butanol was treated with 3 mL of 66% hydrazine, and the mixture was heated on the steam bath for 40 min. The reaction mixture was added to 800 mL dilute H2SO4. The solids were removed by filtration, and the filtrate was washed with 2x75 mL CH2Cl2. The aqueous phase was made basic with 25% NaOH, extracted with 3x75 mL CH2Cl2, and the solvent from these pooled extracts removed under vacuum yielding 6.2 g of a residue that was obviously rich in butanol. This residue was distilled at 138-144 C. at 0.3 mm/Hg to give 0.6 g of a colorless oil. This was dissolved in 2.4 mL IPA, neutralized with concentrated HCl, and diluted with 25 mL anhydrous Et2O. The solution remained clear for about 10 seconds, and then deposited white crystals. These were removed by filtration, washed with additional Et2O, and air dried to give 0.4 g 4-ethoxy-3-ethylthio-5-methoxyphenethylamine hydrochloride (3-TASB) with a mp of 140-141 °C. Anal. (C13H22ClNO2S) C,H. The amber-colored impure phthalimide, following the same procedure, provided another 0.9 g of the hydrochloride salt with a mp of 138-139 °C.
DOSAGE: about 160 mg.
DURATION: 10 - 18 h.
QUALITATIVE COMMENTS: (with 120 mg) This is no more than a plus one, and it didn't really get there until about the third hour. By a couple of hours later, I feel that the mental effects are pretty much dissipated, but there is some real physical residue. Up with some caution.
(with 160 mg) The taste is completely foul. During the first couple of hours, there was a conscious effort to avoid nausea. Then I noticed that people's faces looked like marvelous parodies of themselves and that there was considerable time slowing. There was no desire to eat at all. Between the eighth and twelth hour, the mental things drifted away, but the body was still wound up. Sleep was impossible until about 3:00 AM (the 18th hour of the experiment) and even the next day I was extremely active, anorexic, alert, excited, and plagued with occasional diarrhea. This is certainly a potent stimulant. The next night I felt the tensions drop, and finally got an honest and easy sleep. There is a lot of adrenergic push to this material.
EXTENSIONS AND COMMENTARY: No pharmacological agent has an action that is pure this or pure that. Some pain-killing narcotics can produce reverie and some sedatives can produce paranoia. And just as surely, some psychedelics can produce stimulation. With 3-TASB we may be seeing the shift from sensory effects over to out-and-out stimulation. It would be an interesting challenge to take these polyethylated phenethylamines and assay them strictly for their amphetamine-like action. Sadly, the potencies are by and large so low, that the human animal can't be used, and any sub-human experimental animal would not enable the psychedelic part of the equation to be acknowledged. If an order of magnitude of increased potency could be bought by some minor structural change, this question could be addressed. Maybe as the three-carbon amphetamine homologs, or as the 2,4,5- or 2,4,6- substitution patterns, rather than the 3,4,5-pattern used in this set.
| [ |
[Main Index] | [Forward |