

#138 MPM
2,5-DIMETHOXY-4-(n)-PROPOXYAMPHETAMINE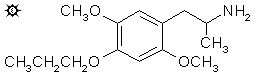
|
| [3D .mol structure] |
To a solution of 3.08 g 2,5-dimethoxyphenol in 20 g MeOH, there was added a solution of 1.26 g flaked KOH in 20 g hot MeOH. There was then added 2.46 g n-propyl bromide, and the mixture held at reflux for 2 h on the steam bath. This was quenched in 5 volumes H2O, made strongly basic with 10% NaOH, and extracted with 3x100 mL CH2Cl2. Removal of the solvent from the pooled extracts left 2.0 g of 1,4-dimethoxy-2-(n)-propoxybenzene as a clear, amber oil. The IR spectrum was appropriate, no phenol was present, and this residue was used in the following reaction without further purification or characterization.
A mixture of 3.5 g N-methylformanilide and 4.0 g POCl3 was held at room temperature for 0.5 h producing a deep red color. To this there was added 2.0 g 1,4-dimethoxy-2-(n)-propoxybenzene, and the mixture was held on the steam bath for 1.75 h. It was then poured over 400 mL shaved ice, and vigorous stirring was maintained until the dark complex had completely broken up. This aqueous mixture was allowed to stand overnight, and the crude aldehyde solids that had formed were removed by filtration, water washed, and sucked as dry as possible. This 2.0 g damp material was crystallized from 20 mL boiling MeOH giving, after filtering and drying to constant weight, 1.4 g 2,5-dimethoxy-4-(n)-propoxybenzaldehyde as reddish-tan solids, with a mp of 97-98 °C. To the methanolic mother liquors of this crystallization there was added a gram of malononitrile and a few drops of triethylamine. The eventual addition of a little H2O encouraged the separation of crystals which were removed, and had a mp of 150-152 °C. Recrystallization from toluene gave gold-colored crystals of the benzalmalononitrile with a mp of 153.5-155 °C, but the melt remained slightly cloudy.
To a solution of 1.4 g 2,5-dimethoxy-4-(n)-propoxybenzaldehyde and 0.65 g nitroethane in 4.4 g glacial acetic acid there was added 0.4 g anhydrous ammonium acetate, and the mixture was heated on the steam bath for 5 h. The addition of a modest amount of H2O and scratching with a glass rod produced crystal seed. The reaction was diluted with about 5 mL H2O, seeded, and allowed to stand at room temperature overnight. There was generated a crystalline product which was removed by filtration and air dried. There was thus obtained 0.6 g 1-(2,5-dimethoxy-4-(n)-propoxyphenyl)-2-nitropropene as yellow-orange crystals, with a mp of 83-84 °C. The addition of H2O to the mother liquors provided an additional 0.3 g of an orange solid which proved to be largely unreacted starting aldehyde.
To a stirred, warm suspension of 0.5 g LAH in 20 mL anhydrous Et2O under a He atmosphere, there was added 0.6 g 1-(2,5-dimethoxy-4-(n)-propoxyphenyl)-2-nitropropene dissolved in a little anhydrous Et2O. The mixture was heated and stirred for a few h, and the excess hydride decomposed with 30 mL 1.5 N H2SO4. The two layers were separated, and 15 g potassium sodium tartrate was dissolved in the aqueous fraction. Aqueous NaOH was then added until the pH was >9, and this was then extracted with 3x50 mL CH2Cl2. Removal of the solvent under vacuum gave 0.7 g of an amber oil that was dissolved in anhydrous Et2O and saturated with anhydrous HCl gas. No crystals formed, and so the ether was removed under vacuum, leaving a residue that set up to crystals that were then no longer soluble in ether. They were, however, very soluble in chloroform. These were ground under dry Et2O, removed by filtration, and air dried giving 0.35 g 2,5-dimethoxy-4-(n)-propoxyamphetamine hydrochloride (MPM) with a mp of 123 - 125 °C.
DOSAGE: 30 mg or more.
DURATION: probably short.
QUALITATIVE COMMENTS: (with 15 mg) This is just barely threshold. A marginal intoxication at best. This level is producing less response that the 11 mg. trial of MEM, so the propoxy is off in potency. At four and a half hours I am out of whatever little there was.
(with 30 mg) By the mid-second hour, I am at a valid plus one. I cannot identify the nature--with eyes closed it would be lost, as it would also be if I were watching a play or movie. It would have been interesting to see where it could have gone. Seventh hour, completely clear.
EXTENSIONS AND COMMENTARY: The 4-propoxy homologue of TMA-2 and MEM is clearly less active, and this has discouraged me from putting too much more effort in this direction. Three additional materials of this pattern were prepared and either shown to be even less active, or simply were not assayed at all. These are the 4-isopropoxy isomer (MIPM), the (n)-butoxy homologue (MBM), and the (n)-amyl homologue (MAM). They scarcely warrant separate recipes as they were all made in a manner similar to this one describing MPM.
For the preparation of MIPM, the above phenol, 2,5-dimethoxyphenol was isopropylated with isopropyl bromide in methanolic KOH giving 2,5-dimethoxy-1-(i)-propoxybenzene as an oil. This formed the benzaldehyde with the standard Vilsmeier conditions, which melted at 77-78 °C from hexane and which gave a yellow malononitrile derivative melting at 171.5-173 °C. The nitrostyrene, from nitroethane in acetic acid was orange colored and melted at 100-101 °C from either methanol or hexane. This was reduced with lithium aluminum hydride in ether to give 2,5-dimethoxy-4-(i)-propoxyamphetamine hydrochloride (MIPM). The properties of the isolated salt were strange (soluble in acetone but not in water) and the microanalysis was low in the carbon value. The molecular structure had a pleasant appeal to it, with a complete reflection symmetry shown by the atoms of the amphetamine side chain and the isopropoxy side chain. But the nature of the actual product in hand had no appeal at all, and no assay was ever started.
For the preparation of MBM, the starting phenol was alkylated to 2-(n)-butoxy-1,4-dimethoxybenzene in methanolic KOH with n-butyl bromide. The benzaldehyde melted at 79.5-81 °C from methanol, and formed a malononitrile derivative that had a melting point of 134.5-135 C. The nitrostyrene from the aldehyde and nitroethane in acetic acid crystallized from methanol with a mp of 71-72 °C. Lithium aluminum hydride reduction in ether gave the ether-insoluble chloroform-soluble product 4-(n)-butoxy-2,5-dimethoxyamphetamine hydrochloride (MBM) with a melting point of 128-130 °C. This product met all tests for structural integrity, and assays were started. At levels of up to 12.0 milligrams, there were no effects noted.
As to the preparation of MAM, the exact same sequence was used, except for the employment of n-amyl bromide. The benzaldehyde crystallized from methanol with a mp of 79-80 °C, and formed a malononitrile derivative which was bright yellow and melted at 103-104 °C. The nitrostyrene, when pure, melted at 57-58.5 °C but proved very difficult to separate from the aldehyde. The final product, 4-(n)-amyl-2,5-dimethoxyamphetamine hydrochloride (MAM) was obtained by lithium aluminum hydride reduction in ether and melted at 125-127 °C. It was assayed at up to 16 milligrams, at which level there was noted a heaviness in the chest and head at the 2-hour point, but no cardiovascular disturbance and no mydriasis. This was called an inactive level, and no higher one has yet been tried.
| [ |
[Main Index] | [Forward |