

#121 MEE
4,5-DIETHOXY-2-METHOXYAMPHETAMINE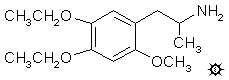
|
| [3D .mol structure] |
A solution of 194 g 3,4-diethoxybenzaldehyde in 600 g glacial acetic acid was arranged in a flask that could be magnetically stirred, yet cooled as needed with an external ice bath. A total of 210 g of 40% peracetic acid in acetic acid was added at a rate such that, with ice cooling, the exothermic reaction never raised the internal temperature above 26 °C. The reaction developed a deep red color during the 2 h needed for the addition. At the end of the reaction the mixture was quenched by the addition of three volumes of H2O, and the remaining acidity was neutralized by the addition of solid Na2CO3 (700 g was required). This aqueous phase was extracted several times with CH2Cl2, and the solvent was removed from the pooled extracts under vacuum. The residue was a mixture of the intermediate formate ester and the end product phenol. This was suspended in 800 mL 10% NaOH, and held on the steam bath for 1.5 h. After cooling, this was washed once with CH2Cl2 (discarded) and then acidified with HCl. There was the formation of an intensely hydrated complex of the product phenol, reminiscent of the problem encountered with 3-ethoxy-4-methoxyphenol. This was worked up in three parts. The entire acidified aqueous phase was extracted with Et2O (3x200 mL) which on evaporation gave 80 g of an oil. The hydrated glob was separately ground up under boiling CH2Cl2 which, on evaporation, gave an additional 30 g of oil, and the aqueous mother liquor from the glob was extracted with 2x200 mL CH2Cl2 which provided, after removal of the solvent, an additional 10 g. These crude phenol fractions were combined and distilled at 1.5 mm/Hg. Following a sizeable forerun, a fraction boiling at 158-160 °C was the anhydrous product, 3,4-diethoxyphenol. It was a clear, amber oil, and weighed 70.0 g. The slightest exposure to H2O, even moist air, give a solid hydrate, with mp of 63-64 °C. This phenol can be used for the synthesis of MEE (this recipe) or for the preparation of EEE (see the separate recipe). A solution of 2.0 g of this phenol in 5 mL CH2Cl2 was diluted with 15 mL hexane. This was treated with 2 g methyl isocyanate followed by a few drops of triethylamine. After about 5 min, white crystals formed of 3,4-diethoxyphenyl-N-methyl carbamate, with a mp of 90-91 °C.
A solution of 26.6 g 3,4-diethoxyphenol in 50 mL MeOH was mixed with another containing 9.6 g KOH pellets dissolved in 200 mL hot MeOH. There was then added 21.4 g methyl iodide, and the mixture was held at reflux for 2 h on the steam bath. This was then quenched in 3 volumes of water, made strongly basic with 25% NaOH, and extracted with 3x150 mL CH2Cl2. Evaporation of the solvent from the pooled extracts gave 19.3 g of 1,2-diethoxy-4-methoxybenzene (3,4-diethoxyanisole) as a clear, pale amber oil that solidified when cooled. The mp was 20-21 °C.
A mixture of 32.0 g N-methyl formanilide and 36.2 g POCl3 was allowed to stand until it was a deep red color (about 0.5 h). To this there was added 18.3 g 1,2-diethoxy-4-methoxybenzene and the exothermic reaction was heated on the steam bath for 2.5 h. This was then poured over 600 mL chipped ice, and the dark oily material slowly began lightening in color and texture. A light oil was formed which, on continued stirring, became crystalline. After the conversion was complete, the solids were removed by filtration producing, after removal of as much H2O as possible by suction, 26.9 g of crude aldehyde. A small sample pressed on a porous plate had a mp of 87.5-88.5 °C. Recrystallization of the entire damp crop from 50 mL boiling MeOH gave, after cooling, filtering, and air drying, 17.7 g of 4,5-diethoxy-2-methoxybenzaldehyde as fluffy, off-white crystals with a mp of 88-88.5 °C. A solution of 1.0 g of this aldehyde and 0.5 g of malononitrile dissolved in warm absolute EtOH was treated with 3 drops triethylamine. There was the immediate formation of crystals which were filtered and air dried to constant weight. The product, 4,5-diethoxy-2-methoxybenzalmalononitrile, was a bright yellow crystalline material, which weighed 1.0 g and had a mp of 156-157 °C.
To a solution of 14.7 g 4,5-diethoxy-2-methoxybenzaldehyde in 46 g glacial acetic acid, there was added 8.0 g nitroethane and 5.0 g anhydrous ammonium acetate. The mixture was heated on the steam bath for 2 h, becoming progressively deeper red in color. The addition of a small amount of H2O to the hot, clear solution produced a slight turbidity, and all was allowed to stand overnight at room temperature. There was deposited a crop of orange crystals that was removed by filtration and air dried. There was obtained 7.0 g 1-(4,5-diethoxy-2-methoxyphenyl)-2-nitropropene as brilliant orange crystals that had a mp of 89-90.5 °C. This was tightened up, but not improved, by trial recrystallization from acetic acid, mp 89-90 °C, and from hexane, mp 90-90.5 °C. Anal. (C14H19NO5) C,H.
To a gently refluxing suspension of 5.0 g LAH in 400 mL anhydrous Et2O under a He atmosphere, there was added 6.5 g 1-(4,5-diethoxy-2-methoxyphenyl)-2-nitropropene by allowing the condensing Et2O to drip into a shunted Soxhlet thimble containing the nitrostyrene. This effectively added a warm saturated solution of the nitrostyrene dropwise. Refluxing was maintained for 5 h, and the reaction mixture was cooled with an external ice bath. The excess hydride was destroyed by the cautious addition of 400 mL of 1.5 N H2SO4. When the aqueous and Et2O layers were finally clear, they were separated, and 100 g of potassium sodium tartrate was dissolved in the aqueous fraction. Aqueous NaOH was then added until the pH was >9, and this was extracted with 3x200 mL CH2Cl2. Removal of the solvent under vacuum produced an off-white oil that was dissolved in anhydrous Et2O and saturated with anhydrous HCl gas. The crystals of 4,5-diethoxy-2-methoxyamphetamine hydrochloride (MEE) that formed were very fine and slow to filter, but finally were isolated as a white powder weighing 5.4 g and melting at 178.5-180 °C. Anal. (C14H24ClNO3) C,H,N.
DOSAGE: greater than 4.6 mg.
DURATION: unknown.
EXTENSIONS AND COMMENTARY: There were early trials made with MEE, before it became known what direction the ethoxy substitution results would take. A number of progressive trials, up to a dosage of 4.6 milligrams, were without any central effects at all.
There is an instinct in structure-activity studies to think of a change as a success or a failure, depending on whether there is an increase or a decrease in the desired activity. But if one were to look at the effects of putting an ethoxy group onto TMA-2 in place of a methoxy group as a way of decreasing the effectiveness, then the 4-position becomes the worst position (MEM is equipotent to TMA-2), and the 5-position is perhaps a little less bad (MME is almost as potent) and the 2-position is the best by far (EMM is out of it, potency-wise). In other words, in the comparison of the 2- and the 5-positions, the lengthening of the 5-position gives modest loss of activity, and the lengthening of the whatever in the 2-position is the most disruptive. With this as a basis for prediction, then MEE (which differs from MEM only by a lengthening of the 5-position substituent) might be only a little less active than MEM and, as MEM is about the same as TMA-2, it is distinctly possible that MEE may show activity in the area at dosages that are not much above the 25 to 50 milligram area. Of all the diethoxy homologues, it would be the most promising one to explore.
Which brings to mind a quotation of a hero of mine, Mark Twain. RI like science because it gives one such a wholesome return of conjecture from such a trifling investment of fact.
| [ |
[Main Index] | [Forward |