

#80 F-22
6-(2-AMINOPROPYL)-2,2-DIMETHYL-5-METHOXY-2,3-DIHYDROBENZOFURAN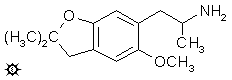
|
| [3D .mol structure] |
In a round-bottomed flask containing an internal thermometer, there was placed 125 g of unpurified 4-(2-methylallyloxy)anisole, and this was heated with an open flame. At an internal temperature of 190 °C an exothermic reaction set in, raising the temperature to 250 °C, where it was held for an additional 2 min. After the reaction mixture had cooled to room temperature, it was poured into 500 mL H2O, made strongly basic with 25% NaOH, and extracted repeatedly with 100 mL portions of CH2Cl2 until the extracts were essentially colorless. These extracts were pooled and the solvent removed to provide 80.0 g of a deeply colored oil that proved to be largely the appropriately substituted dihydrobenzofuran. The aqueous residue from above was acidified with concentrated HCl, and again extracted with CH2Cl2. Removal of the solvent gave 17.7 g of 4-methoxy-2-(2-methylallyl)phenol as an amber oil which eventually set down as white crystals with a mp of 52.5-54 °C.
A solution of 17 g of 4-methoxy-2-(2-methylallyl)phenol in 56 g acetic acid was treated with 8.4 g zinc chloride followed with 28 mL concentrated HCl. This mixture was heated at reflux temperature with a mantle for 1 h. After cooling, this was poured into H2O and extracted with 2x150 mL CH2Cl2. The pooled extracts were washed with several portions of 8% NaOH, until the extracts were colorless. The organic fraction was then washed with H2O, and the solvent removed to yield 5.8 g of 2,2-dimethyl-5-methoxy-2,3-dihydrobenzofuran as a pale amber oil with a pungent smell. This was purified by distillation, giving a fraction of an off-white oil with a bp of 136-138 °C at 33 mm/Hg.
To a mixture of 8.0 g N-methylformanilide and 9.2 g POCl3 which had been allowed to stand for 0.5 h, there was added 4.0 g 2,2-dimethyl-5-methoxy-2,3-dihydrobenzofuran, and the mixture held at the steam bath temperature for 2.5 h. This was then poured into 200 mL H2O which produced a black oily phase that gave no hint of crystallization. This mixture was extracted with 3x150 mL CH2Cl2 and the solvent was removed from the pooled extracts under vacuum. The residual oil (which was shown by GC to contain approximately equal quantities of two isomeric benzaldehydes A and B) was extracted with three 75 mL portions of boiling hexane, each of which on cooling deposited a reddish oil that partially crystallized. A fourth hexane extract gave nothing more. The solvent was decanted from these three extracts, and the semi-solid residues were ground under 3.0 mL MeOH giving 1.4 g of pale yellow crystals of 2,2-dimethyl-6-formyl-5-methoxy-2,3-dihydrobenzo-furan, isomer RBS. After recrystallization from MeOH, the color was almost white, and the mp was 79.5-80.5 °C. The combined mother liquors were enriched in isomer RAS which proved, following preparative GC separation and NMR analysis, to be the 7-formyl isomer. The 80 g of impure dihydrobenzofuran isolated from the Claisen rearrangement above was distilled and a fraction (43.8 g) that boiled from 138-153 °C at 30 mm/Hg was processed as described here to the aldehyde mixture. Following similar hexane extractions, a yield of 4.0 g of a 95% pure isomer RBS was finally obtained. The remaining components of this fraction were not determined, but it is possible that there were some that contained the six-membered benzopyran ring system.
To a solution of 5.2 g of 2,2-dimethyl-6-formyl-5-methoxy-2,3-dihydro-benzofuran in 20 mL glacial acetic acid there was added 3 mL nitroethane followed by 1.6 g anhydrous ammonium acetate. This mixture was heated for 4 h on the steam bath, and then a small amount of H2O was added to the hot solution. This instigated the formation of a copious deposition of brick-red crystals which were, after cooling, removed by filtration, and recrystallized from 50 mL boiling MeOH. After air drying there was thus obtained 2.7 g of day-glo yum-yum orange crystals of 2,2-dimethyl-5-methoxy-6-(2-nitro-1-propenyl)-2,3-dihydrobenzofuran. An additional 0.6 g of product was obtained by working the mother liquors.
A suspension of 2.5 g LAH in 300 mL refluxing anhydrous Et2O was treated with a solution of 3.1 g 2,2-dimethyl-5-methoxy-6-(2-nitro-1-propenyl)-2,3-dihydrobenzofuran in Et2O. The mixture was held at reflux temperature for 18 h. After cooling, the excess hydride was destroyed by the cautious addition of 400 mL H2O which contained 15 g H2SO4. The aqueous phase was separated, washed once with Et2O, and then once with CH2Cl2. There was then added 60 g potassium sodium tartrate, and the pH was brought to above 10 by the addition of 25% NaOH. This was extracted with 3x250 mL CH2Cl2, the extracts pooled, and the solvent removed under vacuum. There remained 2.8 g of an amber oil with an ammoniacal smell. This was dissolved in 200 mL anhydrous Et2O, and saturated with anhydrous HCl gas. There was the immediate formation of an oil, from which the supernatent Et2O was decanted. The residual oil was resuspended in a second 200 mL anhydrous Et2O, again decanted, and finally a third 200 mL Et2O effected the dissolving of the remaining oil to give a clear solution. All three solutions became gelatinous over the following few h, and each deposited a crop of white crystals over the following few days. From the first there was obtained 1.4 g of product with a mp of 153-154 °C; from the second, 0.2 g with a mp of 153-154 °C; and from the third, 1.2 g with a mp of 155-156 °C. These crops were combined, and recrystallized from 10 mL of boiling CH3CN to give 1.7 g 6-(2-aminopropyl)-2,2-dimethyl-5-methoxy-2,3-dihydrobenzofuran hydrochloride (F-22) as a white crystalline solid which had a mp of 154-155 °C. This material, even when dry, showed a tendency to discolor with time.
DOSAGE: greater than 15 mg.
DURATION: unknown.
EXTENSIONS AND COMMENTARY: And here is yet another dihydrobenzofuran which is not of a very high potency if, indeed, it is active at all. This particular dihydrobenzofuran analogue, F-22, had sort of tickled my fancy as being an especially good candidate for activity. It had a certain swing to it. F-22, like LSD-25. And here it was finished, just five days before I had to deliver a paper concerning the syntheses (and activities!) of all these dihydrobenzofurans to the marijuana congress. Could this possibly be another LSD? I was sufficiently convinced that the possibility was real, that I actually started the screening process at a most unusually low level of 10 micrograms. Two days later, I upped this to a dose of 25 micrograms (no activity again) and three days after that, at 1 AM on the polar flight to Copenhagen, I swallowed the "monstrous" dose of 50 micrograms. Shoot the works. If I were to blossom all over the tourist section of the SAS plane, well, it would be quite a paper to give. If not, I could always say something like, "The active level has not yet been found." No activity. Another Walter Mitty fantasy down the tubes.
And, as it turned out, the entire project pretty much ran out of steam. A number of clever analogs had been started, and would have been pursued if there had been any activity promised of any kind with any of these dihydrobenzofurans. The "other" benzaldehyde described above, could have been run in a manner parallel to that proposed for the counterpart with F-2, to make the eventual amphetamine, 7-(2-aminopropyl)-2,2-dimethyl-5-methoxy-2,3-dihydrobenzofuran. Great strides had been made towards F-233 (I have discussed the naming system under F-2, with the F standing for the furan of benzofuran and the 2 and 3 and 3 being the positions of the methyl groups on it). The reaction of 4-methoxyphenol with 1-chloro-3-methyl-2-butene gave the ether which underwent the thermal Claisen rearrangement to 2-(1,1-dimethylallyl)-4-methoxyphenol with a bp of 148-157 °C at 30 mm/Hg. This was cyclized to the intermediate cycle 2,3,3-trimethyl-2,3-dihydrobenzofuran which, after distillation, was shown to be only 80% pure by GC analysis. This was, nonetheless, (and with the hope that is in the very fiber of a young innocent chemist), pushed on to the benzaldehyde stage (and there were a not-too-surprising four benzaldehydes to be found in the oil that was produced, which refused to crystallize). And then (when sheer desperation replaced hope) these were condensed with nitroethane to form an even worse mixture. Maybe something might crystallize from it? Nothing ever did. Junk. Everything was simply put on the shelf where it still rests today, and F-233, 6-(2-aminopropyl)-5-methoxy-2,3,3-trimethyl-2,3-dihydrobenzofuran, remains the stuff of speculation.
And a start towards F-23, 6-(2-aminopropyl)-2,3-dimethyl-5-methoxy-2,3-dihydrobenzofuran, got just as far as the starting ether, when it occurred to me that the final product would have an unprecedented three chiral centers, and so a total of four racemic pairs of diastereoisomers. And then I discovered that the starting allyl halide, crotyl chloride, was only 80% pure, with the remaining 20% being 3-chloro-1-butene. This would have eventually produced a 2-ethyl-analogue, 6-(2-aminopropyl)-2-ethyl-5-methoxy-2,3-dihydrobenzofuran, with its two chiral centers and two more pairs of stereoisomers (not to speak of the need to devise an entirely new coding system). Unless something were to fall into my lap as a crystalline intermediate, the final mess could have had at least six discreet compounds in it, not even considering optical isomers. And I haven't even begun to think of making the six-membered dihydrobenzopyrans which were the THC analogues that presented the rationale that started the whole project in the first place. A recent issue of the Journal of Medicinal Chemistry has just presented an article describing the reaction of 6-methoxytetrahydrobenzopyran with dichloromethyl methyl ether, and approximately equal amounts of all three of the possible isomers were obtained. That would have been the first step towards making the prototypic compound 7-(2-aminopropyl) 6-methoxy-1,2,3,4-tetrahydrobenzopyran. Just as the benzofurans were all named as F-compounds, this, as a benzopyran, would have been a P compound, but P also is used for proscaline, and there would have been some repair-work needed for these codes.
Time to abandon ship. The fact that I had just synthesized and discovered the strange activity of ARIADNE at about this time, made the ship abandonment quite a bit easier to accept.
| [ |
[Main Index] | [Forward |