

#27 2C-G
2,5-DIMETHOXY-3,4-DIMETHYLPHENETHYLAMINE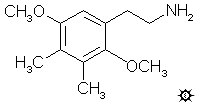
|
| [3D .mol structure] |
A mixture of 205 g POCl3 and 228 g N-methylformanilide was allowed to incubate at room temperature until there was the development of a deep claret color with some spontaneous heating. To this, there was added 70.8 g 2,3-dimethylanisole, and the dark reaction mixture heated on the steam bath for 2.5 h. The product was then poured into 1.7 L H2O, and stirred until there was a spontaneous crystallization. These solids were removed by filtration, H2O washed and air dried to give 77.7 g of crude benzaldehyde as brown crystals. This was distilled at 70-90 °C at 0.4 mm/Hg to give 64.8 g of 2,3-dimethyl-4-methoxybenzaldehyde as a white crystalline product with a mp of 51-52 °C. Recrystallization from MeOH produced an analytical sample with a mp of 55-55.5 °C. Anal. (C10H12O2) C,H. The malononitrile derivative (from the aldehyde and malononitrile in EtOH with a drop of triethylamine) had a mp of 133-133.5 °C from EtOH. Anal. (C13H12N2O) C,H,N. Recently, this aldehyde has become commercially available.
A solution of 32.4 g 2,3-dimethyl-4-methoxybenzaldehyde in 800 mL CH2Cl2 was treated with 58.6 g 85% m-chloroperoxybenzoic acid and held at reflux for 3 days. After cooling to room temperature, the white solids (m-chlorobenzoic acid) were removed by filtration (about 40 g when dry). The filtrate was extracted with several portions of saturated NaHCO3 (on acidification, this aqueous wash yielded additional m-chlorobenzoic acid) and the organic solvent removed under vacuum. The crystalline residue (weighing 32 g and deeply colored) was dissolved in 150 mL boiling MeOH to which there was added 18 g of solid NaOH and the solution heated on the steam bath for a few min. The mixture was added to 800 mL H2O, and a little surface scum mechanically removed with a piece of filter paper. The solution was acidified with concentrated HCl, depositing 30.9 g of a tan solid. Recrystallization from H2O gave 2,3-dimethyl-4-methoxyphenol as white needles, with a mp of 95-96 °C. Anal. (C9H12O2) H; C: calcd, 71.06; found 70.20. The N-methyl carbamate was made by the treatment of a solution of the phenol (1 g in 75 mL hexane with 5 mL CH2Cl2 added) with 2 g methyl isocyanate and a few drops of triethyl amine. The pale pink solids that separated were recrystallized from MeOH to give a product that had a mp of 141-142 °C. Anal. (C11H15NO3) C,H,N.
To a solution of 23.1 g flake KOH in 250 mL hot EtOH there was added 61.8 g 2,3-dimethyl-4-methoxyphenol followed by 60 g methyl iodide. This was held under reflux for 12 h, then stripped of solvent under vacuum. The residue was dissolved in 1.2 L H2O, acidified with HCl, and extracted with 3x200 mL CH2Cl2. The combined extracts were washed with 3x100 mL 5% NaOH, and the solvent was removed under vacuum. The residue set up as an off-white mass of leaflets weighing 37.7 g after filtering and air drying. Recrystallization from MeOH gave 2,3-dimethyl-1,4-dimethoxybenzene as white solids, with a mp of 78-79 °C. Anal. (C10H14O2) C,H. An alternate route leading from 2,3-xylenol to this diether via nitrogen-containing intermediates was explored. The sequence involved the reaction of 2,3-xylenol with nitrous acid (4-nitroso product, mp 184 °C dec.), reduction with sodium dithionite (4-amino product, mp about 175 °C), oxidation with nitric acid (benzoquinone, mp 58 °C), reduction with sodium dithionite (hydro-quinone) and final methylation with methyl iodide. The yields were inferior with this process.
A mixture of 88 g POCl3 and 99 g N-methylformanilide was allowed to incubate until a deep claret color had formed, then it was treated with 36.5 g 2,3-dimethyl-1,4-dimethoxybenzene and heated on the steam bath for 3 h. It was then poured into 1 L H2O, and stirred until the formation of a loose, crumbly, dark crystalline mass was complete. This was removed by filtration, and dissolved in 300 mL CH2Cl2. After washing first with H2O, then with 5% NaOH, and finally with dilute HCl, the solvent was removed under vacuum yielding 39.5 g of a black oil that solidified. This was extracted with 2x300 mL boiling hexane, the extracts were pooled, and the solvent removed under vacuum. The yellowish residue crystallized to give 32.7 g 2,5-dimethoxy-3,4-dimethylbenzaldehyde with a mp of 46-47 °C. Repeated recrystallization from MeOH raised the mp to 59-60 °C. The malononitrile derivative was prepared (aldehyde and malononitrile in EtOH with a few drops triethyl amine) as yellow crystals from EtOH, with a mp of 190-191 °C. Anal. (C14H14N2O2) C,H; N: calcd, 11.56; found, 11.06, 11.04.
To a solution of 16.3 g 2,5-dimethoxy-3,4-dimethylbenzaldehyde in 50 mL nitromethane there was added 3.0 g anhydrous ammonium acetate, and the mixture was heated on the steam bath overnight. There was then added an equal volume of MeOH, and with cooling there was obtained a fine crop of yellow crystals. These were removed by filtration, washed with MeOH, and air dried to provide 4.4 g of 2,5-dimethoxy-3,4-dimethyl-beta-nitrostyrene with a mp of 120-121 °C which was not improved by recrystallization from MeOH (50 mL/g). The mother liquors of the above filtration were diluted with H2O to the point of permanent turbidity, then set aside in a cold box. There was a chunky, granular, tomato-red crystal deposited which weighed 2.5 g when dry. It had a mp of 118-119.5 °C, which was undepressed in mixed mp with the yellow sample. Both forms had identical NMR spectra (2.20, 2.25 CH3; 3.72, 3.84 OCH3; 6.80 ArH; 7.76, 8.28 CH=CH, with 14 cycle splitting), infrared spectra, ultra violet spectra (max. 324 nm with shoulder at 366 nm in EtOH, two peaks at 309 and 355 nm in hexane), and microanalyses. Anal. (C12H15NO4) C,H,N.
A solution of LAH (56 mL of a 1 M solution in THF) was cooled, under He, to 0 °C with an external ice bath. With good stirring there was added 1.52 mL 100% H2SO4 dropwise, to minimize charring. This was followed by the addition of 3.63 g 2,5-dimethoxy-3,4-dimethyl-beta-nitrostyrene in 36 mL anhydrous THF over the course of 1 h. After a few minutes further stirring, the temperature was brought up to a gentle reflux on the steam bath for about 5 min, then all was cooled again to 0 °C. The excess hydride was destroyed by the cautious addition of 9 mL IPA followed by 2.5 mL 15% NaOH and finally 7.5 mL H2O. The reaction mixture was filtered, and the filter cake washed first with THF and then with IPA. The filtrate was stripped of solvent under vacuum and the residue was distilled at 110-120 °C at 0.2 mm/Hg to give 2.07 g of 2,5-dimethoxy-3,4-dimethylphenethylamine as a clear white oil. This was dissolved in 10 mL IPA, neutralized with concentrated HCl, and then diluted with 25 mL anhydrous Et2O. The crystals that formed were filtered, Et2O washed, and air dried to constant weight. There was obtained 2.13 g of beautiful white crystals of 2,5-dimethoxy-3,4-dimethylphenethylamine hydrochloride (2C-G) with a mp of 232-233 °C. Anal. (C12H20ClNO2) C,H.
DOSAGE: 20 - 35 mg.
DURATION: 18 - 30 h.
QUALITATIVE COMMENTS: (with 22 mg) I am completely functional, with writing and answering the telephone, but the coffee really tastes most strange. While the mental effects (to a ++ only) were dispersing, the body still had quite a bit of memory of the day. Sleep was fine, and desirable, in the early evening.
(with 32 mg) Superb material, to be classified as a 'true psychedelic' unless one is publishing, in which case it could be best described as an 'insight-enhancer' and obviously of potential value in psychotherapy (if one would wish to spend 30 hours in a therapy session!). I suppose it would be best to simply stick with the insight-enhancing and skip the psychotherapy. Just too, too long. There was not any particular visual impact, at least for me. The non-sexual and the anorexic aspects might indeed change, with increasing familiarity. Remains to be seen. The length of the experience is against its frequent use, of course, which is a pity, since this one is well worth investigating as often as possible.
(with 32 mg) There was, at the very beginning, a certain feeling of non-physical heat in the upper back which reminded me of the onset of various indoles, which this ain't. The energy tremor was quite strong throughout, but somehow the body was generally at ease.
(with 32 mg) At a plateau at two hours, with just a bit of tummy queasi-ness. And I am still at the plateau several hours later. Sleep finally at the 18th hour, but even after getting up and doing all kinds of things the next day, I was not completely baseline until that evening. And a couple of days more for what is certainly complete repair. That is a lot of mileage for a small amount of material.
EXTENSIONS AND COMMENTARY: Here is the first example, ever, of a phen-ethylamine that is of about the same potency as therelated three-carbon amphetamine. At first approximation, one is hard put to distinguish, from the recorded notes, any major differences either in potency, in duration, or in the nature of activity, between 2C-G and GANESHA itself.
I had always thought of the phenethylamines as being somewhat weaker than the corresponding amphetamines. Sometimes a little weaker and sometimes a lot weaker. But that is a totally prejudiced point of view, an outgrowth of my earliest comparisons of mescaline and TMA. That's the kind of thing that can color one's thinking and obscure what may be valuable observations. It is equally valid to think of the phenethylamines as the prototypes, and that the amphetamines are somewhat stronger than the corresponding phenethylamines. Sometimes a little stronger and sometimes a lot stronger. Then the question suddenly shifts from asking what is different about the phenethylamines, to what is different about the amphetamines? It is simply a historic fact, that in most of my exploring, the amphetamine was made and evaluated first, and so tended to slip into the role of the prototype. In any case, here the two potencies converge.
| [ |
[Main Index] | [Forward |