

The End of a Chemistry Era...
Dave Nichols Closes Shop
November 2012
Citation: Hanna J, Manning T. "The End of a Chemistry Era.... Dave Nichols Closes Shop". Erowid Extracts. November 2012;23:2-7.

|
By using newly synthesized chemicals and known compounds as "molecular probes", Nichols has explored how psychoactives interact with the structures and systems of neurons and how changes in neurochemistry affect behavior. His laboratory has published dozens of studies elucidating details of the mechanism of action of MDMA and of the biochemical events related to its neurotoxic effects as seen in animals.
Nichols coined the term "entactogen" as a descriptor for the unique psychopharmacological effects of MDMA and similar drugs. He is an expert on the pharmacology and chemistry of these compounds, as well as LSD, mescaline, and their analogs. He has contributed to more than two hundred scientific papers on related topics.
Nichols is President of the Heffter Research Institute, a nonprofit organization that he co-founded in 1993 to promote research with psychedelics "in order to contribute a greater understanding of the mind leading to the improvement of the human condition, and to alleviate suffering."
In June of 2012, Nichols retired as professor of Medicinal Chemistry and Pharmacology at Purdue and relocated to the University of North Carolina at Chapel Hill. Erowid jumped at the opportunity to speak with him before the move--and before his lab was dismantled. Although Nichols is well known for his work with psychedelic compounds that affect serotonin receptors, we were intrigued to learn that he has also deeply researched medicinal substances that affect dopamine.
Dave Nichols: Aside from hallucinogens, I have a parallel research track, which focuses on dopamine agonists. With a colleague named Richard Mailman, who was at the University of North Carolina at Chapel Hill, I co-founded a small biotech company called DarPharma. We developed drugs, first in class, for Parkinson's disease, and that may be useful in improving working memory in schizophrenia, as well. We've got six or seven patents now, all in that field, because no one has paid any attention to it. One of the drugs, dihydrexidine, is in clinical trials now. We were going to commercialize it; initially we were looking at Parkinson's disease, although that market is fairly small. It's kind of an interesting situation; since there is no drug out there marketed that has this kind of pharmacology, nobody knew exactly what to do. We eventually hired a new CEO, and she said, "Why aren't you looking at schizophrenia as an indication, because the market is a lot bigger?"
So we paid to have a market analysis done, and it was something like a $10 billion dollar per year market for this, for improving memory and cognition in schizophrenia. That study is going on now at Columbia University. If it works, that will be a proof of principle. We've already proved it works, in monkeys, for Parkinson's disease. It's better than anything that's out there. And if this works in schizophrenia, this would be a game changer. But also, I have all the patents--so it probably would help me in my retirement a little bit. [Nichols smiles]
Jon Hanna: Could it also be useful as a nootropic for normals?
Dave: We don't know anything about what it would do there. It's never been tested for that kind of indication. There have been studies on drug abuse that showed these sorts of compounds block cocaine craving. And if it improves working memory and cognition in schizophrenia... In the monkeys, they've shown that it improves it in aged monkeys. My guess is that people my age or older could probably take this thing. It's a weird compound--these kinds of compounds--because, in monkeys, you only have to treat them a couple of times, and the effect lasts a year. The doses are 10-4 to 10-5 mg/kg--it's like 10 µg/kg of monkey weight. This is a drug that maybe, as a human, you would take 100 micrograms every other day for a week, and then, your memory will be back for a year. That would be the most optimistic scenario. Everybody I run into that's past the age of about 40 or 50 says, "I would love to have the memory I had when I was younger." So, if it really worked there, you are looking at a market with a size that is unimaginable. But I'm basically just keeping my fingers crossed for this first clinical trial in schizophrenia.
|


Because I worked in the dopamine area, as well as with serotonin [5-hydroxy-tryptamine, or 5-HT], I could see a lot of things. We did the first pharmacology of MDMA in 1982, showing that it was a serotonin releaser. Then we did work on the so-called neurotoxicity and showed there's an interaction between dopamine and serotonin. I could see a lot of that because I also worked with dopamine, and I knew a great deal about dopamine agonists, amphetamine, and so forth.
My funding for research on hallucinogens and dopamine started at almost the same time and ended at almost the same time. I had 28 years of funding for both of those projects. So, really, they're two separate research areas. Being in two different fields made me conversant with things that people, if they just worked in one area, wouldn't have seen.
Tania Manning: In 2010, at the Psychedelic Science conference, you mentioned how around the fifth hour of an LSD trip, many people experience a change in the tone of its effects. Tell us a bit about your research into this.
Dave: Yes, right. Danny Freedman was one of the early clinical investigators of LSD when he was at the University of Chicago. I knew him; we were at meetings together and had written some things together. I would see him, and he'd say, "You know Dave, when we gave LSD to people, the first four or five hours it was very psychedelic and wonderful, and then for most people, it would change at about the mid-point. It would get very dark, and some people would get paranoid ideation, and ideas of reference, and things that looked a lot like amphetamine psychosis--paranoid psychosis." He said, "Nobody's ever studied that change in effects, and I think it's important to look at."
Danny's remarks stuck in my mind for a long time. Because we'd worked on MDMA, and knew that activation of serotonin receptors then stimulated dopamine biosynthesis and turnover, I thought, "Well, I wonder if there's something there..." We had done some other research, within mostly the amphetamine/MDMA field, and I started thinking, "I wonder if we could see a change in the effects produced by LSD in rats?" Normally we would give rats LSD, and then we would train them 30 minutes later, once they were under the influence of the LSD, to press a lever to get a food pellet. They learned to do that reliably. They learn to recognize the effect of LSD, and once they feel that, they know that they can press a lever and get food. And if you try to use different antagonists to block the effect, 5-HT2A receptor antagonists will block the LSD effect and the rats won't press the lever. I suggested to my research associate, Danuta Marona-Lewicka, "Why don't we wait 90 minutes? Give LSD and wait 90 minutes before we train them?"
Because we'd worked on MDMA, and knew that activation of serotonin receptors then stimulated dopamine biosynthesis and turnover, I thought, "Well, I wonder if there's something there..."
The dopamine hypothesis of schizophrenia is one that's held sway for a long time. It posits that excess dopaminergic activity is responsible for some of the symptoms of schizophrenia, and--in particular--psychosis. You give amphetamine, or psychostimulants, or cocaine to people in large doses for a long period of time, and they become psychotic. So all of a sudden, LSD brought in this dopamine effect. I thought, "Well, maybe there's something happening there."
We published probably half-a-dozen papers on that. Actually, my son is following that up. When these rats are given LSD chronically, they start developing very weird behaviors, which are very much like models of schizophrenia in animals. We proposed that this might be an animal model of schizophrenia. The people who are heavily invested in the current models didn't like it too well. But my son is doing a lot of genetics stuff now; he was trained in Drosophila [fly] genetics. He sees changes in gene expression in these rats that parallel some of the things that he's seen in post-mortem schizophrenic brains. In fact, today, he just asked me, "What weight rat should I use? I'm going to start doing this and look at it." The idea is that this might be a model for schizophrenia. The best drugs to treat schizophrenia block both dopamine D2 receptors and 5-HT2A receptors. LSD activates D2 receptors and 5-HT2A receptors. So it kinda made sense. Then the question was, if you had a dopaminergic effect, what could be the basis for that? It turns out that, at least in rats, if you pretreat them with something like either DOI or LSD, and you wait two hours--Danuta did a time course, and we did this in rats that were trained to recognize the effects of amphetamine--their response to amphetamine is increased; the potency of amphetamine increases. What this means is, pretreatment with a 5-HT2A agonist causes a delayed sensitization of dopamine systems; that's the way that we interpreted it. It's consistent with what happens with MDMA--the way it turns on dopamine by activating 5-HT2A receptors. So that would fit.
Then there was a PET study published in Synapse in 2005, where the researchers administered a PET ligand that bound to dopamine D2 receptors in pigs. Then they gave LSD to the pigs. Normally you give an animal a positron-emitting ligand drug, in this case it was a D2 receptor blocker, and then you intravenously give the drug that you expect to displace it, and you see whether it was displaced, and then you follow its disappearance from the brain. Usually if you give pigs LSD, a very high concentration would be there immediately and you would see immediate displacement of the radioactive ligand, and then you would see a long-term, slower displacing effect. What these researchers observed was, when they gave LSD to these pigs, they did not see maximal receptor occupancy for four hours. They concluded that could mean either that the dopamine system had become sensitized over time, which is exactly what we'd been thinking, or maybe there's an active metabolite of LSD, which is also what we'd been thinking. You can't find anything about the metabolites of LSD in the literature from anything more recent than, say, the 1970s. But back then, researchers had identified 13-hydroxy-LSD as one of the human metabolites of LSD. They just identified it, that's all. If it's a metabolite of LSD, well, metabolites are produced in the liver. And you have 15 or 20 isoforms of these mixed-function oxidases that can put hydroxy groups onto compounds during metabolism. It varies, depending on you. Ethnic groups have different levels of them, there are different mixtures, etc. Depending on what your P450s were, if that isozyme was dominant, it might be producing a lot of 13-hydroxy-LSD; if it wasn't dominant, it may be that you don't produce much of it. So then there's a genetic biological basis for explaining why there would be variability, if the change in LSD's effect is actually due to an active metabolite.
I was interested in the possibility that 13-hydroxy-LSD was an active metabolite that interacted with the dopamine system, because the fact that some people were having psychosis-like symptoms during the latter part of their LSD trips might point us to a direction in terms of understanding schizophrenia.
Jon: You haven't published yet regarding your speculation that the change in LSD's effects after about five hours might be due to an active metabolite, right?
Dave: Right. I could publish something like that in Medical Hypothesis. We've given posters where we have said that it's been postulated that there may be an active metabolite. But we haven't really explicitly outlined what we think might be going on.

|
Dave: No. Ralf Heim had published a paper, years ago, on some of these compounds. I was intrigued by them, and we probably made 20 or 25 of them, to map out the SAR [structure-activity relationship] and find out what was going on. He'd just made the compounds and never really did any SAR studies. Heim had studied several of these N-benzyl phenethylamines. We just fleshed it out more, and probably got a lot more publicity, because his thesis and the publication were in German. We published in the journal Molecular Pharmacology, so our work was more accessible. The NBOMes are super-potent compounds. Initially, I didn't think they were orally active.
Tania: Switching gears to the Iodo compounds, like DOI, did you mention that these are anti-inflammatory?
Dave: Yeah, that's very weird.
Tania: Some guy called in to Sasha's office, saying that he had rheumatoid arthritis, and he took 2C-I, and for two weeks he was pain-free. Which makes me wonder about medical applications for the Iodo compounds...
Dave: My son Chuck discovered that accidentally. He's an associate professor at Louisiana State University in New Orleans. He wanted to work with 5-HT2 agonists, because he's looking at serotonin receptors in Drosophila, and doing translational stuff into rats. He asked, "Is there a 5-HT2A agonist that's not a controlled substance that I can use?" Since DOI was not controlled, I sent him the isomers of DOI.
His team had been using rat aortic epithelial cells--cells from the inside of a rat's blood vessels--and looking at models of atherosclerosis. The model they'd been using was to take these cells, and put in TNF-alpha (tumor necrosis factor-alpha), a pro-inflammatory substance. If you've seen the advertisements for Enbrel, for arthritis, drugs such as that block TNF-alpha receptors, so they block the pain. What they would do is put TNF-alpha directly into these cells and then they would look at what effect occurred in combination with other compounds--there were four or five compounds that they were looking at.
So his post-doc had some of those cells that were grown up and could be used, and he asked my son, "What if I run a test with one of our compounds in these?" And Chuck said, "Well, I don't have any anti-inflammatory compounds right now." "What about this DOI here?" Chuck laughed and replied, "That's a hallucinogen. That won't do anything." The post-doc said, "Well, I'm going to have to destroy the cells. Can I just go ahead and test it?" And Chuck said, "Yeah, go ahead." The guy came back a week and a half later and said, "The DOI completely blocked TNF-alpha at 20 picomolar." Which is like unbelievable, right?
Chuck said, "Nah. You made a mistake." So Chuck went in, made up his own fresh solutions, took the cells, ran the experiment, and reproduced the guy's data. He wrote me back and asked, "Is there any precedent for this?" And I said, "No, not that I know of." So he published a paper in J PET; it was the featured paper in the issue it was published in. This has extraordinary potency; there's no anti-inflammatory that has potency like that.
Jon: The dose levels you mentioned would not be psychoactive, so perhaps that's something that could be developed into a commercial medication.
Dave: Exactly, exactly. I've sent him a bunch of isomers. But so far he hasn't found anything that's as potent as DOI. And the thing is, the affinity of DOI at the human receptor is like one nanomolar, or around there; so at the concentrations he's using, the amount of receptor that's actually occupied has to be incredibly small. There is some mechanism here that nobody really understands; and that was a big controversy when he sent the paper in. People were like, "What is this?"

|
Dave: Yeah. That's really the only reason. In fact, I have a paper that I'm getting ready to send in--we're just waiting for one more piece of data--where we've made some analogs of DOI due to our anticipating that DOI is going to eventually become scheduled. And DOI is a really valuable research tool if you want to study the 5-HT2 system. There have been almost 600 papers published that used DOI. If DOI is actually scheduled and people can't use it, this is basically going to be a disaster. So we've got one analog that Tocris Bioscience sells, TCB-2, which is also a 5-HT2A agonist. It's never been compared with DOI, but it does kind of the same things. Our new compounds are cyclopropane compounds that look fairly similar to DOI and DOB. We're going to publish on this and suggest that maybe these can "fill in" if DOI is scheduled.
Jon: What pharmacology or chemistry do you feel most proud of having come from your team at Purdue?
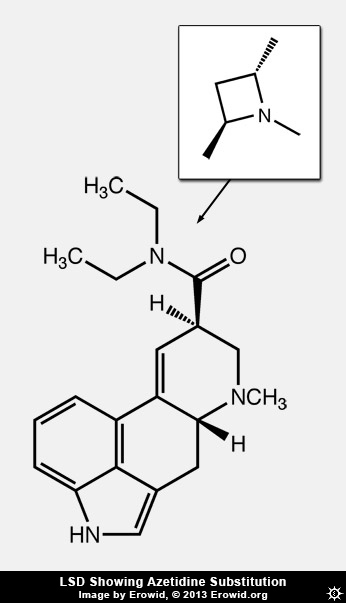
Dave: I wouldn't say it's pharmacology, but one of the cutest things we did was to make azetidine analogs of LSD: using the diethylamide, we took a carbon and hooked the two ethyls of LSD together and that gave us a dimethylazetidine. We had the methyls on the same side of that four-membered ring or on the opposite sides, either with R,R or S,S stereochemistry, showing that it was just one of those that was most like LSD. This proved that with LSD, when the diethylamide binds, those diethyl groups bind in a specific way. Then we modeled it, and we did mutagenesis of the receptor--and we haven't published this yet--and found that leucine 229 in extracellular loop 2 was the thing that interacted with that diethyl amide. The fact that the receptor has evolved specifically to be complementary to LSD, that is interesting. [Jon intonates Twilight Zone theme music.] It took us about ten years to figure out how to do that.
First we tried to make an aziridine, which is a three-membered ring. I had a student, Rob Oberlender, who worked on that for a long time. Those were unstable--the ring just broke open. So we went to the four-membered azetidine rings. In medicinal chemistry when you go from methyl to ethyl or propyl or things like that, you don't expect to see the activity change this dramatically, unless there's a unique binding site or there is some steric restriction binding site. With LSD you change it, and it's different. We pretty much knew there had to be some place in the receptor where that was binding. But to actually be able to demonstrate it, and then show that the ethyl groups bind in this specific orientation, and then do the mutagenesis which basically proves that--that was kind of cool.
|
I don't think many people even know that my team supplied these drugs. They might know that Dave Nichols is "this chemist." But where did the materials come from for the so-called psychedelic renaissance that is occurring?
Rick Doblin called me and said, "I can't find anybody to make MDMA. Can you guys do it?" "Yeah, I can do that." I made a kilo and a half, and MAPS didn't need it all, so we donated it; most of the clinical studies have been done with stuff that I made back in 1986. Strassman, as he says, was the first in a generation to give a psychedelic to humans. I worked closely with him. He said, "Dave, what if I get all these approvals and I can't get the DMT?" I said, "Well, I made the MDMA for Doblin. I can probably make DMT for you." That's exactly what happened. He got to the end and he said, "Nobody wants to make the DMT." So I made the DMT.
Then Roland Griffiths needed some psilocybin, so I was asked, "Can you make psilocybin?" Stewart Frescas and I worked together to improve the synthesis of psilocybin, and that process has been used to make all the psilocybin for the clinical studies. I'm pleased with that contribution. It doesn't look like a major thing, but having psilocybin available at an affordable cost for all these clinical studies has really opened that whole area up, by figuring out a better synthesis and getting the material out to clinical investigators. We spent a couple of years figuring out the best way to make it, then making that first batch of four grams that Roland used in the spirituality study. So that's great. That's a great paper. Even though I didn't discover psilocybin, making it available was a good thing.
To some extent, I'm kind of living vicariously through other people's clinical work, knowing that I contributed the material. And now, our synthesis of psilocybin is available. Other people have told me that they sent our improved synthesis method to more people. So it's out there.
Eventually somebody will do a study with LSD. We didn't do it at the Heffter Institute, just because there was still so much social stigma, and it's a long-acting compound. We discussed what we should use when we were going to treat terminal patients. We went with psilocybin because it was more benign, shorter-acting, and nobody knew what it was.
Jon: The Heffter Research Institute's well-considered, cautious decision regarding what material was used with that cancer anxiety study laid some solid stepping stones...
Dave: Heffter is where I've been able to do the clinical stuff, which I couldn't otherwise have done because I don't have an MD. Working with Franz Vollenweider... with the research he's doing with brain imaging, he's helping unravel the mystery of consciousness itself. That's the big question: What is consciousness? Because if we're not conscious, we're not here. You can't really understand altered states of consciousness, unless you understand consciousness. That's tied up in a lot of what Franz is doing, but also a lot of what Heffter is doing.
We may treat terminal patients, because that's the first indication and that's where we have the biggest benefit-to-risk ratio--in cancer patients. But I think we are going to find things there that are going to lead us into other areas. People who aren't dying are also sometimes anxious and depressed. So then you can say, "Okay, let's do a clinical study and look at those people." I think we're going to open up some of these other avenues. For me, it's just the beginning.
Tania: How does your work with Heffter tie into what you'll be doing at Chapel Hill?
Dave: The people at Heffter are hoping that I'll have more time to work for them after I've retired. If I was going to go into this field today, I might wonder, really, how many more ligands do we need?
I made a bunch of them. Sasha made a bunch of them. Other people have made some. But I don't know what all of those tell you, at this point.
We have lots and lots of tools. What's missing is how to connect those tools with how they produce altered states of consciousness. And there, I think you need more clinical stuff, more PET scans, more EEG, functional MRI, magneto-encephalography, which nobody has used with a psychedelic at all; you actually get the brain currents. And it's all tied up with the nature of consciousness.
What is consciousness? What produces consciousness? Those are big issues. Psychedelics could be really useful tools in understanding that.
Jon: Changing gears, what is your favorite psychoactive plant or chemical?
Dave: [laughs] I'll refuse to answer that.
Jon: Well, it might be coffee, or chocolate, or wine! And certainly there was a time not that long ago when consumption of many of the entactogens scheduled these days was not illegal. How many times, for example, have you taken MDMA, MDAI, MDE, or MBDB?
Dave: Before they were scheduled?
I think that if you're going to have a drug as an entactogenic psychotherapeutic agent, it ought to be one that drug-naive people could take without having any kind of an overwhelming experience.
Dave: If you would think that, then you'd probably be thinking correctly. But I suspect I'm much less experienced than people might imagine that I am. I am conservative by nature, and this is a conservative place. Set and setting are not really conducive. When I initially started, if I had a strong feeling that something was going to be interesting and it was a new substance, well... bioassays were the tradition in chemistry. So certainly I had some feedback at times on compounds where it seemed indicated that this is worth looking at, and that other things weren't worth looking at.
Tania: Considering all of the years that you studied the neurotoxicity of entactogens, is there one drug that stands out, other than MDMA, that might be suitable for therapeutic use?
Dave: As an entactogen, probably MBDB. I'm still intrigued by the possibility that it could be useful as an adjunct to psychotherapy. For people who experimented with MDMA before it was a controlled substance, the thing that they said was that with MDMA, there's this grandiose euphoria that hits you. And MBDB doesn't really produce that.
With MBDB, there have been people who have taken it who have said, "You know, I didn't really notice when it started. It was just that all of a sudden, I realized that I was in, and had been in, this altered state of consciousness."
I think that if you're going to have a drug as an entactogenic psychotherapeutic agent, it ought to be one that drug-naive people could take without having any kind of an overwhelming experience.