

10 Neuropharmacology of DXM
used at Erowid with the permission of author William White10.1 What is a Receptor, Anyway? (Basic Neuropharmacology)
10.1.1 The Structure of a Nerve Cell
All cells have a voltage gradient between the outside and the inside of the cell; if you were to measure the voltage between the inside and outside of the cell you would find that the inside of the cell is somewhat less than -100mV compared to the outside of the cell. In most cells this charge is a consequence of the pumping of ions into and out of the cell, and is not used for anything in particular. Muscle and nerve cells, however, make use of this charge.
Muscle cells and nerve cells (neurons) have "excitable" membranes -- that is, the charge across the cell membrane can change, and this change is used for specific purposes. In neurons, a drop in this charge is used to convey a signal.
There are more than 10 billion nerve cells in the human brain, and they all do the same job: conveying signals from their inputs (dendrites) to outputs (axons). A nerve cell looks a little bit like a tree, except that instead of roots and branches, a nerve cell has dendrites and axons. Generally speaking, the dendrites receive signals and the axons transmit them (remember, a signal is indicated by a change in cell membrane voltage).
At the end of each branch of the axon is a synaptic bouton, a small button-like structure which is used to send a signal to the next nerve cell (more on that later). Conversely, the dendrites are used to receive signals, and contain numerous little bumps, knobs, and such that each receive a signal from another neuron. One neuron may receive signals from thousands of other neurons.
Some of the signals may excite the cell membrane; others may inhibit it. If the sum of excitatory minus inhibitory signals received by the dendrites is large enough, the signal will reach the nerve cell body (soma). Once a strong enough signal is received by the soma, it is sent out along the axons and transmitted.
This is the basis for how neurons function individually. However, it doesn't explain how one neuron sends signals to another (or conversely, how they receive signals). A very few neurons are physically joined to each other, and signals simply cross from one cell to another just as the signals from one transistor cross to another inside a microchip. However, the majority of neurons don't touch each other; instead, they communicate with chemicals called neurotransmitters and receive them with structures called neuroreceptors
10.1.2 Neurotransmission
A receptor is a structure on the surface of, or inside, a cell. If on a nerve cell (neuron), it is often called a neuroreceptor. Receptors exist to receive signals from particular chemicals, and when they receive these signals they generally exert some sort of change on the function of the cell. Some receptors are present inside the cell and are called intracellular receptors; many receptors for steroids (testosterone, estrogen, etc.) are intracellular.
Since this chapter is about neuropharmacology, the focus will be on neuroreceptors, usually referred to herein simply as receptors. Neuroreceptors exist on the surface of (or, more rarely, inside) nerve cells and respond to chemicals called neurotransmitters. Some neurotransmitters also act on cells other than nerve cells; for example, acetylcholine activates receptors on muscle cells, telling them to contract.
Neurotransmitters fit into receptors like keys into a lock, and do not, in general, fit into any other receptor. Thus we have acetylcholine, a neurotransmitter, and acetylcholine receptors. Some receptors (called ion channel receptors) when activated will stimulate or depress the membrane potential (and thus the nerve cell's activity); others (metabotropic receptors) will induce changes in in the characteristics of the cell. Some ion channel receptors (calcium channels) can do both.
Generally speaking, a receptor on a nerve cell is positioned so that it can receive signals from another nerve cell. Dendrites of course have receptors, but receptors can also exist on the cell body, on axons, or on synaptic boutons. The "interface" between the two nerve cells is called a synapse.
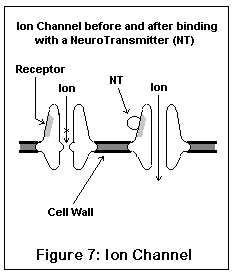
The ion channel neuroreceptors typically operate very quickly, and act (and look) somewhat like an iris shutter in a camera. The neurotransmitter (for example, acetylcholine) binds to a specific area on the channel, which (due to electrostatic forces) causes the channel to snap open. Specific ions then leak into and out of the nerve cell, changing its electrical potential. Different channels allow different ions to pass; some ions (like sodium) excite the nerve cell, others (like potassium and chloride) inhibit it. Once the neurotransmitter leaves the receptor, the channel snaps shut, having done its work. These are the receptors involved in fast signal transmission, and in conveying skeletal muscle impulses.
The metabotropic receptors have a modulatory role. Some of them increase or decrease the number of other types of receptors. Some cause changes in genetic expression in the cell. Some (called autoreceptors) inhibit the release of their own matching neurotransmitter, a process called negative feedback. A thermostat is an example of a negative feedback system -- the hotter it gets, the less the furnace is on. Generally, these slower domain receptors operate by second messengers (which function as messengers within the cell) such as G-proteins.
Any given neurotransmitter will probably be associated with several different receptors. For example, serotonin (5HT) activates at least twelve receptor subtypes (5HT1A, 5HT1B, 5HT1D, 5HT1E, 5HT1F, 5HT2A, 5HT2C, 5HT3, 5HT4, 5HT5, 5HT6, and 5HT7)! There are several subtypes (instead of just one) because each receptor subtype is involved in a different process on a different type of neuron.
Drugs act on the brain by affecting neurotransmission in some way or another. Some drugs stimulate receptors, some block them; some will change the way that neurotransmitters are secreted, degraded, or recycled. To a great degree, drugs work only because they affect existing neurotransmitter systems; in spite of the popular belief that getting high equates to frying brain cells, drugs that make you high do so simply by mimicking, blocking, or otherwise affecting neurotransmission.
Drugs which mimic, block, or otherwise affect activity of a given neurotransmitter will not affect all receptor subtypes equally. For example, LSD operates at 5HT2A and 5HT2C receptors; buspirone operates at 5HT1A receptors. Consequently, they have very different effects; LSD is psychedelic, whereas buspirone is an anti-anxiety drug.
Different substances may bind to the same receptor but affect it differently. An agonist is a substance which binds to the receptor and activates it. A partial agonist is an agonist which does not activate the receptor fully. An antagonist binds to the receptor and prevents it from operating.
One interesting property of partial agonists is that they tend to "normalize" receptor activity levels. In the presence of a low amount of neurotransmitter, the partial agonist will increase receptor function. In the presence of a high amount of neurotransmitter, however, the partial agonist will limit receptor activity; in fact, many antagonists may really be partial agonists. It is still being debated as to whether LSD is a 5HT2C antagonist or a partial agonist.
Antagonists may bind to the same place where the neurotransmitter binds, thus "competing" with the neurotransmitter - these are called competitive antagonists. Or they may bind to a separate place on the receptor complex, so that even if the neurotransmitter reaches its binding site, the receptor won't activate. These are called noncompetitive antagonists. Note that in either case, the binding of the drug is only temporary; if it were permanent (thus effectively destroying the receptor) it would be irreversible antagonism.
The important difference between a competitive and a noncompetitive antagonist is this. If you block receptors with a competitive antagonist, these receptors can still be activated by neurotransmitters if enough neurotransmitter is secreted. If you block a receptor with a noncompetitive antagonist, however, no amount of neurotransmitter will activate that receptor (until the noncompetitive antagonist goes away).
A rather whimsical analogy can be made between neurotransmitter functioning and toilets. In this case, the toilet is the receptor, you are the neurotransmitter, activating it by pushing the flush handle. If your little brother comes up and flushes the toilet for you, he's is an agonist. If he temporarily sticks the handle halfway down, he's a partial agonist. If he holds the handle up so it won't flush, he's a competitive antagonist. If he plugs up the toilet with toilet paper, he's a noncompetitive antagonist. If he breaks the toilet handle off completely, he's an irreversible antagonist.
The biogenic amine neurotransmitters (so called because they are in a chemical class called amines) include acetylcholine, noradrenaline, dopamine, serotonin (5HT), and histamine. They are derived from amino acids (choline, tyrosine, tyrosine, tryptophan, and histidine respectively), generally have a modulatory role, and are the common targets of recreational drugs. For example: LSD, DMT, and psilocybin target 5HT receptors; amphetamine causes a release of dopamine and noradrenaline; cocaine blocks the reuptake of dopamine (thus keeping it active longer); MDMA causes a release of 5HT and dopamine; etc. A mostly complete list of recreational drugs and their neuroreceptor activity is given in Section 15.2.
The neuropeptide neurotransmitters include a whole slew of peptides (chains of amino acids), such as neuropeptide Y, angiotensin, endorphins, substance P, and so on. The only recreational drugs targeting neuropeptide receptors are the opiates, which target the mu, kappa, and delta opioid receptors. Opioid receptors are (obviously) involved in pain and behavioural reinforcement. Vasopressin, a nootropic ("Smart Drug") is also a peptide neurotransmitter.
The amino acid neurotransmitters include GABA (gamma-aminobutyric acid), glutamate, and aspartate. Receptors for these neurotransmitters include the GABA receptors (which come in two main flavors) for GABA, and the NMDA, AMPA (formerly quisqualate), kainate, and metabotropic receptors (all of which respond to glutamate and aspartate). The GABA receptor is the target of benzodiazepines like diazepam (ValiumTM), barbiturates, and alcohol; the NMDA receptor is targeted by PCP, ketamine, alcohol, and DXM.
And then there are those receptors that don't really fit in anywhere else. The anandamine receptor is the recently-identified target for the THC in marijuana. The adenosine receptor, which tends to inhibit nerve activity, is blocked by caffeine (by which it exerts its stimulant effect). The sigma receptor was originally classified as an opioid receptor, but is now thought to be separate. Gamma-hydroxybutyrate, GHB, seems to target a specific receptor as well.
Each receptor can have more than one binding site (a place where a drug can bind to, generally affecting the activity of the receptor). For example, the NMDA channel/receptor complex has seven (glutamate, glycine, magnesium ion, zinc ion, PCP open channel site, polyamine site, and phosphorylation site). Most have fewer than this; the NMDA channel is an extremely complicated receptor.
Voltage Dependent Ion Channels are similar to the fast-domain, shutter-like receptors, except that they are opened by voltage potentials across the cell membrane. They usually transmit signals along nerve fibers, or cause the end of an axon to release its neurotransmitter. Sodium, potassium, calcium, and chloride (Na+, K+, Ca2+, and Cl-) are the usual ions in question. Tetrodotoxin, the active ingredient in "zombie powder", is a sodium channel blocker. The NMDA receptor has some features of a voltage dependent ion channel (see below).
10.2 What are Sigma Receptors?
Discovered in 1976, sigma receptors (sigma is often written in Greek -- σ -- and if your web browser accepted that, you're luckier than I am) are currently one of the most confusing entities in neuropharmacology. Our knowledge of sigma receptors pales in comparison to our ignorance; in fact, what we absolutely know (or at least think we absolutely know) can be summed up very briefly in the following paragraph:
Scattered throughout the brain and body there are places (sigma binding sites) where a bunch of chemicals (sigma ligands) happen to stick. We don't know if they're on the outside or inside of cells. We don't know if sticking a chemical to them does anything or not, except in the vas deferens. We don't really know what they do, if they do anything. We don't know what they're for, why they're there, or whether the body uses them. They may be neuroreceptors, steroid receptors, intracellular messenger receptors, growth regulators, enzymes, or something else entirely.
In other words, prepare to be confused. Don't worry, everyone else is as well.
Sigma receptors were originally thought of as opioid receptors, since many morphine derivatives bind there (283). However, this classification is probably false, and the endogenous opioid peptides show little sigma activity. The usual characteristics of opiates are mediated by the mu (μ), kappa (κ), and delta (δ) receptors. There are at least two sigma receptors, and a third one (sigma3, appropriately enough) has been discovered recently (114).
Some researchers have speculated that sigma receptors aren't really receptors at all, but just enzyme binding sites (84). On the other hand, sigma ligands affect the guinea pig vas deferens muscles, which probably wouldn't happen unless sigma receptors really were receptors (97). Sigma receptors may be intended for hormones or intracellular messengers rather than neurotransmitters, as they are present on microsomes rather than on the cell surface (129).
10.2.1 Sigma 1 Receptors and General Sigma Information
Much of what is known about sigma receptors seems to apply more to sigma1 than sigma2 (though this is by no means universal). I grouped the following information with sigma1 receptors, but don't take this as gospel. I expect that a lot will turn out to be wrong. Fortunately, we may not have to wait long; research in sigma receptors is proceeding rapidly.
10.2.1.1 Endogenous Ligands
The neurotransmitter for sigma1 receptors has not been found, although there are speculations and evidence (82-86,98-99). The usual term for the (unidentified) sigma1 neurotransmitter is "endopsychosin" (99), formerly known as "angeldustin". Progesterone targets sigma1 receptors in the placenta, and it and other steroid hormones may be natural ligands for sigma1 receptors (97,102,103). If this is true, it is possible that some of the effects of sex hormones on the brain may be mediated by the sigma1 receptor (97). Substance P (a peptide neurotransmitter) was considered but rejected as an endogenous sigma1 ligand (111). DHEA may be a sigma1 agonist, and progesterone an antagonist (258).
10.2.1.2 Location and Function in the Brain
Sigma receptors are densest in the cerebellar cortex (281), nucleus accumbens, and cortex, and also present at lower density in the limbic areas and extrapyramidal motor system (260). This is interesting because some of the bizarre effects of DXM on motion may be related to sigma activity in the cerebellar cortex and extrapyramidal motor system.
Sigma1 receptors (and possibly sigma2) appear to be functionally coupled to some other receptors, notably nicotinic acetylcholine receptors (97,116) and NMDA receptors (106-109,275). They may actually be located on or near NMDA receptors (222).
The nicotinic receptor coupling may be direct, with sigma activation causing a change in the function of nicotinic receptors. Whether modulation of nicotinic receptors would alter the effects of nicotine on the brain, I don't know; some people have indicated that tobacco induces strong responses during DXM use.
Sigma agonists (and/or possibly antagonists) seem to affect memory function, reversing the impairment in memory caused by drugs such as p-chloroamphetamine and MK-801 (a drug similar to ketamine) (130,131). DTG, (+)-pentazocine, and SKF-10047 all improved memory impairment caused by MK-801. On the other hand, NE-100, which is considered a sigma antagonist, seems to help with NMDA antagonist induced memory impairment as well (106-107). DTG, a sigma agonist, reversed the memory impairment caused by carbon monoxide (117).
Many drugs now considered sigma antagonists or agonists may in fact be partial agonists. Another possibility is that the optimal level of sigma activity may be a healthy medium; one study found a bell-curve dose response on sigma agonists (117). This is similar to the effect of many nootropics (smart drugs), specifically the cholinergics - taking too much can be worse than taking none at all. This similarity may be further evidence for the link between sigma receptors and acetylcholine receptors.
Both sigma1 agonists and antagonists may protect NMDA receptors from glutamate toxicity (108). One study found that sigma antagonists protected hippocampal cells from hypoxia and hypoglycemia (104), and this may be related to NMDA receptors as well. Morphine has indirect effect on NMDA receptors that seems to be mediated via sigma receptors, probably sigma1 (109). It is possible that all these effects are mediated via the nicotinic receptor, i.e., sigma1 may not directly control NMDA functioning.
10.2.1.3 Behavioural Effects
The behavioral effects of sigma1 receptors have not been fully established. However, sigma1 (and sigma2) receptors seem to have effects on motor function, producing an increase in locomotion (112,113,120). Part of this effect may occur at the cerebellum (112); the release of dopamine may also be involved (113,128). This is probably the origin of DXM's curious effects on motions and gait, including "sea legs" and the "Robo Shuffle".
Sigma1 activation may counteract some of the analgesic effects of opioids (118). Pentazocine (Talwin), a synthetic opiate, is a potent sigma1 agonist which tends to be self-limiting; when too much is taken, the sigma activity reverses the opiate activity (267). It is possible that the gradual loss of euphoric effects experienced by morphine and heroin users may be related to changes caused by sigma activity.
Sigma receptors seem to be involved in psychotomimetic (literally "psychosis-like") effects from schizophrenia and drugs (46-49). Amphetamine psychosis, a temporary condition resulting from heavy use of psychostimulants, may be due in part to sigma1 activity (80,125). Sigma, and in particular sigma1, receptors may be altered by schizophrenia. An alternative possibility which is being studied is that some sort of chemical - produced by the body itself, or by a virus or other foreign agent - causes prolonged activation of sigma receptors, and this is one of the causes for schizophrenia (47,49). Many neuroleptics, including some of the atypical ones, are sigma antagonists (47,260). Some antidepressants are also sigma antagonists, and all eventually reduce sigma binding (260). Methamphetamine increases sigma binding (211).
Sigma receptors may be involved in the function of the pineal gland, an endocrine gland which secretes melatonin (which maintains the "biological clock"). Sigma activity modulates the noradrenaline-stimulated synthesis of melatonin in the pineal gland (256), and there are in fact sigma receptors within the pineal gland (266).
In addition to DXM, other recreational drugs such as PCP, cocaine, and opiates all show activity at sigma receptors (72). Chronic amphetamine use increases the number of sigma receptors (80), while chronic antidepressant and antipsychotic treatments decrease the number of sigma receptors (47,74). Sigma receptors are involved in the limbic areas of the brain (81) and thus may be involved in emotion. They are also involved in the cough reflex, and probably involved in seizures (or at least their prevention).
10.2.1.4 Location and Function in the Body
Sigma1 receptors are also present throughout the body. Most tumor cells express both sigma1 and sigma2 receptors (38,105), and sigma agonists can inhibit tumor growth (269). Liver and kidney cells also contain sigma receptors (123), as do heart cells (124), and splenocytes (274). As stated above, the placenta contains sigma1 receptors.
Sigma receptors are also present in the immune system and endocrine glands, and may be responsible for modulating these systems. Sigma agonists can prevent rejection of grafted tissue by regulating T cells (263). There is some evidence that sigma agonists may inhibit the immune system. The widespread presence of sigma receptors may indicate some involvement in development, cellular regulation, or other basic biological process.
10.2.2 Sigma 2 Receptors
Much of what was stated about sigma1 receptors may apply to sigma2 receptors as well. There hasn't been much time to differentiate between the two receptor types. The neurotransmitter for sigma2 receptors may be zinc ions (78), and sigma2 receptors seem related to potassium ion channels (79). The sigma2 receptor is less affected by DXM than the sigma1 receptor (58). Some of the sigma-induced potentiation of NMDA function may be due to sigma2 receptors (116). Ibogaine, currently being tested as a cure for heroin addiction, is a sigma2 agonist (273,283). Other ligands for the sigma2 receptor have been found (261,265,268).
One study found that chronic exposure to sigma ligands, both agonists and antagonists, caused brain cells to degenerate and die (101). The deterioration occurred as a gradual loss of cellular shape; cells eventually became spherical (and died soon after). Interestingly, some drugs, including DXM, seemed to be very weak in this effect. While haloperidol induced significant changes and cell death in a few hours, it took DXM 3 days to produce any changes at all, which reversed when the DXM was removed. The potency of different sigma ligands seems to point towards sigma2 receptors as the culprit in this effect.
By the way, I wouldn't worry too much about this. The concentration of DXM required to induce any change at all was extremely high, and it took 3 days of constant exposure. All changes were reversible, even after the cells had assumed a spherical shape. Haloperidol and other sigma ligands, which seem to be up to 100 times as potent as DXM at producing brain cell degeneration, are used medically used without substantial evidence of brain damage. Finally, steroid hormones may very well cause the same sort of effects if present at sufficient levels (another reason not to use anabolic steroids, I guess).
10.2.3 Sigma 3 Receptors
Sigma3 receptors are a new discovery (114). They seem to be linked to the conversion of tyrosine to dopamine, and sigma3 agonists may increase the rate of dopamine synthesis. DXM's potency at the sigma3 receptor is unknown, but if it binds strongly there, then increased dopamine synthesis may be partially responsible for DXM's stimulant effects. A few sigma3 ligands have been found (262).
10.3 What Are NMDA Receptors?
10.3.1 NMDA and Other Glutamate Receptors
Most of the better known neurotransmitter systems - dopamine, noradrenaline, serotonin (5HT), and acetylcholine in particular - have modulatory roles. They are produced by a few neurons located in specific clusters, and drugs affecting them often have specific effects (recreational or medical, or both). Receptors for these neurotransmitters tend to operate fairly slowly, taking milliseconds or longer to communicate. Rather than directly changing the potential of the neuron, they often trigger second-messenger responses.
On the other hand, most of the brain's regular function operates quickly, and involves the excitatory and inhibitory amino acids (EAAs and IAAs, respectively). The receptors for amino acids are generally ion channels; when the receptor is activated, ions enter or exit the cell which change its potential. EAA and IAA synapses generally correspond to the positive and negative synaptic connections in electronic and computer neural networks.
The excitatory amino acid neurotransmitters include glutamate and aspartate. GABA is the only established inhibitory amino acid neurotransmitter in the brain; the spinal column also uses glycine. Generally, glutamate is more prominent (or at least better understood) than aspartate, although they have similar effects at EAA receptors. Thus, the receptors for EAAs are called glutamate receptors.
There are currently four identified type of glutamate receptors. Two of them, the AMPA (formerly quisqualate) and kainate receptors, are ion channel receptors which increase neuron activity in response to EAAs. A third, the metabotropic glutamate receptor, is a newer discovery, and seems to involve second messenger systems and produce metabolic effects. The fourth is the NMDA receptor.
10.3.2 NMDA Receptor Structure and Function
This drawing represents the structure of the NMDA receptor, according to current knowledge.
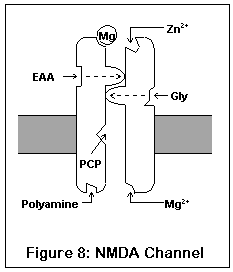
The NMDA receptor has seven distinct binding sites. Three of these are located on the exterior surface of the cell, two are located on the cell interior, one on the inside of the channel, and one (the magnesium ion site) is present both on the inside and outside surfaces.
There are two agonist sites on the exterior are the cell, denoted EAA and Gly; they correspond to the excitatory amino acids (glutamate and aspartate) and glycine. Both sites must be occupied before the channel can open enough for any ions to pass through. A third site is the target of zinc ions (Zn2+), which block the channel when present.
The exterior of the channel contains a magnesium ion site. This site is also present on the inside of the cell (alternatively, it may be located within the channel itself). A magnesium ion normally occupies the exterior site; the interior site is probably empty under biological conditions.
The interior of the cell contains two binding sites. One binds to polyamines (spermine and spermidine), and its function is unknown. The other, not shown in this diagram, is a phosphorylation site. Enzymes can bind to this site and enhance or reduce the activity of the receptor.
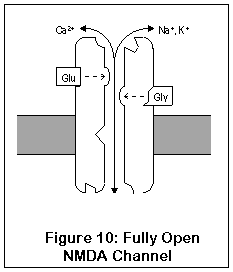
Finally, inside the channel itself is the PCP1 site, where PCP, ketamine, MK-801 (dizocilpine), DXM, and dextrorphan all bind. The channel must be fully open for these drugs to enter; once in place they "clog up" the channel.
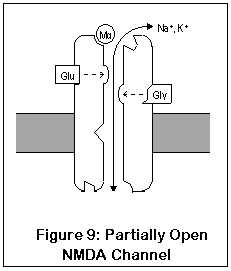
NMDA receptors are unique for several reasons. Unlike most receptors, they require two agonists (glutamate or aspartate, plus glycine) before the channel opens. These two agonists (Glu and Gly in the diagram) bind to two different locations on the NMDA receptor. After both agonists have bound to the channel, it opens enough for potassium to enter, and the receptor operates much like AMPA and kainate receptors. This is shown in Figure 9.
The most important and unique characteristic of NMDA receptors, though, is what happens next (Figure 10). Normally, a magnesium ion is bound to a specific location at the opening of the channel; this ion allows potassium to pass through but prevents calcium, possibly due to its size. This binding is due to electrostatic forces.
Once the cell becomes activated enough, however, the cell potential rises enough that the magnesium ion is no longer stuck to the cell. Calcium can enter (and exit, although this doesn't happen) the cell through the fully open NMDA channel. Once inside, calcium sets into motion a series of responses which enhance the strength of the synapse.
So what's the point? Well, if the neuron is only slightly active, the NMDA channel may open partially, but the magnesium ion won't get a chance to leave its binding site. However, if the neuron should be rapidly or substantially activated, the magnesium ion will be released, and calcium can enter the cell, enhancing synaptic strength. This process of enhancing synpatic strength, called Long-Term Potentiation (LTP), is one of the mechanisms by which neurons can change their functioning and "learn". LTP in the hippocampus is probably responsible for short-term memory. Learning capacity may in fact be directly related to the number of NMDA receptors in the hippocampus (where intermediate-term memory is thought to be stored) (88). LTP is reversible, and long-term memory seems to be stored via more permanent changes in genetic expression and synaptic shape.
Another interesting aspect to the dual action of the NMDA receptor is that it functions as a sort of "coincidence detector". Calcium only enters the cell when the membrane potential is low enough and when the synapse is activated; thus it detects when synaptic and intracellular signals coincide. In associative networks (which the hippocampus seems to be), the NMDA receptor is ideally constructed to help a neuron "learn" an association between two input signals.
There are at least three types of NMDA receptors (in the rat, at least; this probably extends to humans as well). One type is found in the cerebellum, one in the thalamus, and one in the cortex. These types differ subtly, but it is possible that DXM may show a different spectrum of effect on these types than other NMDA antagonists (such as ketamine or PCP) (87). There is also some speculation that the NMDA receptor's ion channel may (for reasons unknown) become "uncoupled" from the receptor itself (63).
Noncompetitive antagonism of NMDA receptors by the open channel blockers is known to induce changes throughout the brain. NMDA blockade causes an increase in dopamine release in the midbrain and prefrontal cortex (63). NMDA blockade also causes activation of 5HT systems specifically targeting the 5HT1A receptor (90).
10.3.3 NMDA Receptors and Excitotoxicity
NMDA receptors are involved in excitotoxicity (nerve cell death via over-stimulation). The chemicals which agonize (activate) NMDA receptors can also kill the very same nerve cells they are activating (19). Many substances, such as quinolinic acid (a metabolite of tryptophan) are so potent that very small amounts can devastate great numbers nerve cells. Others, like glutamic and aspartic acid, are less potent but still capable of doing damage if present in sufficient amounts. This excitotoxicity is directly responsible for much of the damage attributed to various types of trauma and insult to the CNS. Polio is a good example; by blocking the activity of quinolinic acid, all the damage resulting from poliomyelitis can be prevented (30-31). DXM is not a particularly effective NMDA open channel blocker, but DXO, PCP, ketamine, and MK-801 (dizocilpine) are all very effective blockers.
Unfortunately, nothing in life is ever free. Lowered NMDA activity, called Olney's Lesions, NMDA Receptor Hypofunction (NRH), or NMDA Antagonist Neurotoxicity (NAN) seems to be itself responsible for excitotoxicity to other neurons. The theory is that normal NMDA activity keeps other neurotransmitters (glutamate and acetylcholine, and possibly dopamine) from being over-secreted. NMDA blockade releases this inhibition, and can therefore lead to hyperactivity at some neurons. It is possible that chronic NMDA blockade may be a cause for, or at least a factor in, schizophrenia and Alzheimer's disease (100).
NMDA blockade at recreational levels has not been well studied, and Olney's lesions appear only at doses far in excess of recreational levels. My hunch is that occasional NMDA blockade is probably not terribly traumatic to the brain; otherwise, John Lilly would be a lot dumber than he is. DXM in particular may be safer due to its lower potency at NMDA receptors and possible counteracting effects of sigma activity. For more information see Section 6.3.1.
One final note: infants may be particularly susceptible to this effect, so use of any NMDA antagonist during pregnancy or nursing is probably a bad idea (112). It has even been suggested that fetal alcohol syndrome is mediated in part by the effects of NMDA blockade.
10.4 What are PCP2 Receptors?
PCP2 receptors were, obviously, the second PCP receptor to be positively identified (the first is the open channel site on the NMDA receptor). Their use by the body (if they have one) has not been determined. Most research indicates that the PCP2 receptor is the dopamine reuptake complex, the very same one targeted by cocaine and methylphenidate (RitalinTM) and possibly by the antidepressant bupropion (WellbutrinTM) (70,126).
A reuptake complex (or reuptake site), incidentally, is a structure on a cell which takes used neurotransmitter back into the cell for recycling or breakdown. By blocking reuptake of a neurotransmitter, its activity can be increased. The tricyclic antidepressants block the reuptake of noradrenaline, dopamine, and/or serotonin (5HT). Fluoxetine (ProzacTM) is a serotonin-specific reuptake inhibitor (SSRI), as are several other newer antidepressants. The dopamine reuptake site seems to be the only reuptake site targeted by recreational drugs (primarily cocaine).
Curiously, bupropion, a dopamine reuptake inhibitor, seems to have little recreational use potential; then again, it isn't a particularly strong dopamine reuptake inhibitor. It has been suggested that it is a competitive reuptake inhibitor, so that it is only capable of inhibiting reuptake of small amounts of dopamine. Large amounts of dopamine will overwhelm the effects of the competitive inhibition. about
10.5 What are Na+ and Ca2+ Channels?
Sodium and calcium ion channels are two types of voltage dependent ion channels. These channels open or close not due to neurotransmitters, but instead due to voltage differences between the inside and outside of the cell.
Voltage dependent sodium channels are typically involved in the action potential - a domino-effect propagation of nerve impulses along the axon. The sodium channel opens when the voltage reaches a certain activation threshold; the resulting influx of sodium then further activates the neuron (leading to more sodium channels opening). Eventually a second part of the sodium channel closes (otherwise they would keep themselves open forever). Incidentally, voltage dependent potassium channels are involved in bringing the neuron back to its resting state.
Voltage dependent calcium channels are similar to voltage dependent sodium channels, and typically open on activation voltages. Their effect, however, is to cause calcium to enter the cell; the calcium then acts as a messenger to intracellular mechanisms. The most common example is at the end of the axon, where calcium influx causes neurotransmitters to be released. NMDA receptors may be structurally related to voltage dependent calcium channels.
DXM has been found to block sodium and calcium channels, although it is not particularly potent in this capacity. Because of their extensive presence, blockade of these ion channels could have overall depressant effect upon brain function, and might explain DXM's toxic effects at very high dosages.
10.6 How Does DXM Compare to Other Dissociatives at These Receptors?
PCP and ketamine both bind more strongly to NMDA, and less strongly to the PCP2 and sigma sites, than DXM. In fact, some users report that DXM, at higher dosages, begins to resemble ketamine and PCP. The resemblance is still fairly limited. DXM's unique characteristics are most likely due to the PCP2 and sigma sites.
10.7 Endopsychosin and the Big Picture
For whatever reason, some people involved in biological sciences like to talk about the "big picture." I'm one of them. I think the reason why the "big picture" seems so important is that science, especially biological science, has become so specialized and compartmentalized that it's difficult to keep one's perspective, especially when considering the possible relevance of things.
Endopsychosin (en-doe-sy-KOE-sin) is the name given to an endogenous ligand for the NMDA open channel site (PCP1) and/or sigma receptors. The search for endopsychosins started several years ago in an attempt to find the endogenous ligand for PCP; at the time, the term was "angeldustin". Recently, the search for endopsychosins has resumed as NMDA and sigma receptors have become increasingly understood. As I write this, nobody has managed to positively identify an endopsychosin, although there are several candidates. The most promising candidates for the NMDA PCP1 site seem to be series of peptides (98-99). The endogenous ligand for the sigma1 site may be an unknown aromatic chemical (97,99).
The original idea behind endopsychosin (or angeldustin, if you prefer) was that the body was capable of secreting a substance which would mimic the effects of PCP on the brain. It may be secreted in times of extreme stress, leading to a sort of detached, dreamy feeling. Endopsychosin may be responsible for such altered states of consciousness as religious ecstasy, speaking in tongues, possession, astral projection, and other paranormal experiences. Spontaneous releases of endopsychosin may account for experiences such as alien abductions, encounters with ghosts, and that sort of thing. On the other hand, these states might be better explained by temporal lobe complex partial seizures, or other electrical mishaps.
Note the similarity of these experiences with aspects of DXM, ketamine, and PCP drug trips. In particular, the "emergence phenomenon" identified with ketamine (and present also with PCP and DXM) often consists of experiences with spiritual or alien beings.
What's going on here? Why the hell would the human brain secrete a chemical that makes us think we've been talking to Elvis and Jim Morrison on the far side of Mars? What's the big picture?
Well, to be honest, nobody knows. One potential clue is that the perforant path of the hippocampus (a neural circuit) seems to release endopsychosin when stimulated (138). Perhaps endopsychosin is a part of the memory process; or perhaps it is involved in dreaming and the conversion of intermediate-term to long-term memories. Another suggestion is that endopsychosin is involved in long-term depression (LTD), the flip side of long-term potentiation.
Another possibility is that endopsychosin is one of the brain's natural defenses against injury. I find it interesting that sigma/NMDA agents often mimic fever hallucinations; common characteristics include Lilliputian hallucinations, geometrical and linear hallucinations, and dysphoria. Perhaps the brain secretes endopsychosins during high fever in an attempt to prevent neurotoxicity.
In addition to potential neuroprotective roles, these substances may have significant roles in regulating cognition and (in the body) the immune and endocrine systems. A dysfunction of an endopsychosin, or of the sigma receptors (or both) may be one of the causes of schizophrenia. And if some steroids (e.g., progesterone and testosterone) turn out to be endopsychosins, this could explain a lot about the long-term behavioral effects of steroid use.
Or, it may simply be that altered states of consciousness are a natural part of animal life, and that our culture's fear of such states is abnormal. Certainly one doesn't need drugs to achieve altered states; even profoundly dissociative states can be achieved with a certain amount of ritual and faith. Most "primitive" cultures have some experience with dissociative states such as astral projection, shamanic journeying, possession, and that sort of thing. They may very well know something that we don't.
Finally, the perceived effects of endopsychosins (and the drugs that mimic them) may simply be side effects of something more fundamental going on in the limbic system. Perhaps these alien and spiritual encounters and other paranormal phenomena (see Section 8) are the way that the conscious mind interprets limbic network states that normally don't make it to consciousness.
So it is entirely possible that the similarity between NMDA PCP1 and sigma receptors has a purpose. In any case, data about the effects of sigma-specific agonists (or antagonists for that matter) are limited, but our understanding of these receptors should improve in the next few years as research continues. Not to mention the possibility of some brave and/or stupid psychonaut deciding to experiment with sigma-specific agonists.
(+)-3-PPP and SKF-10,047 are good sigma-specific ligands; more sigma1 specific ligands include 1-phenylcycloalkanecarboxylic acid derivatives (122,127) Anyone feeling brave? Maybe you can become the next Shulgin ("Endopsychosins I Have Known And Loved" anyone?). Then again, maybe you'd better not; I don't need to be sued if you develop a stubborn case of insanity.

| 
| 
|