

The Neuropharmacology of Ethyl Alcohol
v1.0, Aug 2005
Citation: Bilz0r. "The Neuropharmacology of Ethyl Alcohol". Erowid.org. July 2005; v 1.0. Erowid.org/chemicals/alcohol/alcohol_pharmacology1.shtml
Introduction #
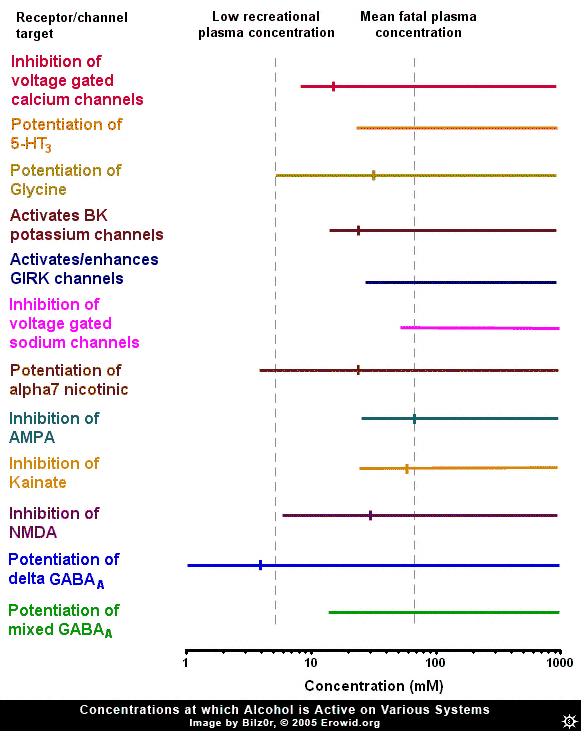
Ethanol (alcohol, ethyl alcohol) is the world's most commonly used recreational drug, yet, despite many years of research, ethanol's exact pharmacological mechanism remains somewhat elusive. Ethanol has been shown to affect a member of nearly every type of ion channel in the body, but often at concentrations far above those found in recreational users. In order to make sense of the huge amount of research into the effects of ethanol on neurons, one must gauge it against the threshold plasma concentration of ethanol needed to produce an effect and a rough estimation of the fatal concentration in humans, i.e. the range of ethanol's concentration found during recreational usage. It is estimated that the plasma concentration necessary to cause threshold effects in humans is 5 mM (40 mg/100 mL) (Rang et al., 2001) while in a review of 808 fatal alcohol poisonings, the mean plasma concentration was 72 mM (Koski et al., 2002). To put blood concentrations further in context, the blood-alcohol limit in most states in the US is 0.08% wt/vol or 17.4 mM.GABA-A #
Because of ethanol's depressive effects, there has been constant speculation that its site of action is the GABA-A receptor (the major inhibitory receptor in the brain, the same receptor enhanced by benzodiazepines and barbiturates). Many experiments have looked into this possibility, and they broadly fit into two categories: ones that found no effect and ones that found a relatively high potency effect, well within recreational concentrations. Exactly why there is this stark dichotomy is unclear, though it may have something to do with more recently discovered facts about the distribution of "subunits" of GABA-A receptors.GABA-A Subunits #
The GABA-A receptor is not homogenous throughout the brain, it is a protein made up of 5 subunits, selected from a possible 16 different subunits, named alpha 1 to 6, beta 1 to 3, gamma 1 to 3, delta, epsilon, theta and pi. Delta-subunit-containing GABA-A receptors are relatively rare, making up about 5% of GABA-A receptors throughout the brain, found primarily in the granule cells of the denate gyrus of the hippocampus and the granule cells of the cerebellum. It has been shown that ethanol potentiates the activity of delta-subunit-containing GABA-A receptors with a very high potency (0.1-3mM). It could be that experiments showing ethanol has a high potency effect somehow inadvertently targeted the delta-subunit-containing GABA-A receptors and caused them to activate more than in other experiments. The high potency of this effect, however, might discount delta-subunit containing GABA-A receptors as the target of ethanol's effects, because at plasma concentrations which cause a maximum effect at delta-subunit-containing GABA-A receptors, most people would be barely feeling any effect of ethanol. Also, the relatively low and restricted expression of delta-subunit containing GABA-A receptors throughout the brain makes it hard to explain how ethanol can have such a wide variety of effects, if this specific GABA-A receptor was its site of action.NMDA #
Inhibition of NMDA receptors by ethanol is another theme that has received a lot of attention, because of the similar anaesthetic nature of ethanol and the classical NMDA receptor antagonists, the dissociative anaesthetics (ketamine and PCP). The NMDA receptor is a compelling site for explaining ethanol's actions, as the concentrations of ethanol that start to cause a significant inhibition of the NMDA receptor are the same concentrations at which the effects of ethanol are beginning to be registered in humans (~5 mM). NMDA receptor antagonists are classically known for their ability to block the formation of memories, an effect that larger amounts of ethanol are renowned for. However, one problem with this theory is the lack of similarity between alcohol's intoxication state and that induced by ketamine or PCP.Other Glutamate Receptors #
Besides the NMDA receptor, other glutamate receptors are affected by ethanol. The AMPA and kainate receptors are both inhibited by ethanol, but generally at concentrations above those needed to induce intoxication. On the other hand, the concentration needed to produce the 50% of maximal inhibition (IC50) of AMPA and kainate receptors is approximately the same as the mean fatal concentration of ethanol (~70 mM), indicating that the inhibition of AMPA or kainate receptors could be the cause of ethanol-induced fatalities. This makes sense, as large scale AMPA inhibition is almost certainly fatal. However, it has been demonstrated that there is a small population of kainate receptors expressed on interneurons (small, GABA-releasing neurons) in the hippocampus which are exquisitely sensitive to ethanol (IC50 = ~5 mM). Because of the crossover between the concentration needed to inhibit these receptors, they seem a possible target for ethanol. But again, because this effect is localized to a small area of the brain and is activated by a lower concentration of ethanol than is needed to induce intoxication, they might not be the cause of the major effects of ethanol.
Nicotinic Acetylcholine Receptors #
The nicotinic acetylcholine receptor is another interesting target of ethanol. Ethanol potentiates the alpha7 subtype of the nicotinic acetylcholine receptor at concentrations that overlap the concentration needed to intoxicate. The alpha7 subtype is one of the most common nicotinic acetylcholine receptor subtypes in the brain, the other common subtype, the alpha4beta2 subtype, is not affected by ethanol. It is almost certain that ethanol's classical "depressant" effects are not mediated by this receptor, as the classical nicotinic acetylcholine receptor agonist nicotine is not depressant. However, one effect that nicotine and ethanol share is their addictive nature. It has been demonstrated that nicotinic acetylcholine receptor antagonists inhibit the increase in dopamine release in the brain caused by ethanol (Blomqvist et al., 1997) and the rewarding effects in humans (Blomqvist et al., 2002).Voltage-Gated Sodium Channels #
Ethanol has been shown to inhibit voltage-gated sodium channels (the same channel local anesthetics inhibit), but only at very high concentrations.Potassium Channels and GIRKS #
It has been demonstrated that ethanol modulates potassium channels. Potassium channels are very important in reducing neuronal excitability. The G-protein coupled inward rectifying (GIRK) channel is a channel modulated by CB1-cannabinoid and Mu-opioid receptors (as well as many others). Ethanol has been shown to directly open (or at least potentiate the opening of) this channel at concentrations starting at ~20-75 mM. While this puts the GIRK channel outside the range for mediating ethanol's recreational effects, it could be responsible for some of the effects of ethanol seen at higher doses, such as analgesia. In mice lacking a functional GIRK channel, ethanol had severely reduced potency as an analgesic (Kobayashi et al., 1999). Ethanol has also been shown to enhance the activity of calcium-activated potassium (BK) channels. These channels become active when a neuron fires repetitively, slowing and eventually stopping the neuron from firing. Ethanol activates these receptors in the high range of ethanol blood concentrations, and so may be responsible for some of the depressant effects of ethanol at high concentrations.Glycine Receptor #
The glycine receptor is an inhibitory receptor distributed largely in the spinal cord and brainstem. Ethanol potentiates this receptor at concentrations directly overlaying its recreational plasma concentrations. This could cause a variety of effects like inhibition of signaling from the brain to the body, and might also be responsible for respiratory depression at high doses, but is unlikely to mediate the classical cognitive of ethanol.Voltage-Gated Calcium Channels #
The effect of ethanol on voltage-gated calcium channels is one effect that has received a lot of press. Ethanol inhibits these channels at concentrations very similar to its recreational plasma concentration and has an IC50 in the middle of the recreational range. The opening of voltage-gated calcium channels is responsible for the release of neurotransmitters throughout the body and, by inhibiting this, ethanol will decrease neurotransmitter release. This inhibition causes a decrease in both excitatory and inhibitory synaptic transmission.Serotonin 5-HT3 Activation #
Ethanol has also been shown to potentiate the serotonin 5-HT3 receptor. This receptor is somewhat enigmatic, but is believed to modulate excitation in some parts of the brain, and to facilitate vomiting. The ethanol concentrations that are needed to potentiate the 5-HT3 receptor are outside the range reached during recreational usage.Summary #
Despite ethyl alcohol being one of the most widely used psychoactives, the pharmacology of its altering effects is still not yet fully understood. There are many systems that it is known to affect, but many of the documented neuro-physiological effects only occur at plasma concentrations outside the range that ethanol is used in humans.Its effects on GABA-A receptors seem tantalizing, but generally unlikely, due to either a far too high or too low potency, depending on the subunit makeup. Ethanol's inhibition of NMDA receptors seems likely to be a contributor to its effects. The inhibition of AMPA and kainate receptors is too weak to mediate recreational effects, but it may be responsible for ethanol-induced coma. Ethanol's potentiation of alpha7 nicotinic acetylcholine receptors seems a likely cause of some of its rewarding effects, but not a mediator of its general "depressant" character. Ethanol's effects at voltage-gated sodium channels are far too weak to have any effect during recreational usage. BK channel activation might cause some of the effects of ethanol at high concentrations and GIRK channel potentiation could be responsible for its analgesic effects. Potentiation of glycine receptors might cause spinal inhibition and lead to some effects of ethanol, but not the classical cognitive effects. 5-HT3 receptor effects are too weak to mediate ethanol's effects. Inhibition of voltage-gated calcium channels is another likely mediator of ethanol's effects, as it would cause general inhibition of synaptic activity.
| Collection of Evidence for Ethyl Alcohol's Activity on Various Neuropharmacological Systems | |
|---|---|
| Target of Activity | Evidence of Activity |
| Mixed GABA receptor | |
| Delta containing GABA receptor | |
| NMDA receptor | |
| Kainate receptor | |
| AMPA receptor | |
| Nicotinic acetylcholine receptor | |
| Voltage gated sodium channels | |
| Potassium channels | |
| Glycine receptor | |
| Voltage gated calcium channels | |
| 5-HT3 receptor | |
References #
- Aguayo LG, Pancetti FC, Klein RL, Harris RA. "Differential effects of GABAergic ligands in mouse and rat hippocampal neurons." Brain Res. 1994;647:97-105.
- Blomqvist O, Ericson M, Engel JA, Soderpalm B. "Accumbal dopamine overflow after ethanol: localization of the antagonizing effect of mecamylamine." Eur J Pharmacol. 1997;334:149-56.
- Blomqvist O, Hernandez-Avila CA, Van Kirk J, Rose JE, Kranzler HR. "Mecamylamine modifies the pharmacokinetics and reinforcing effects of alcohol." Alcohol Clin Exp Res. 2002;26:326-31.
- Carta M, Ariwodola OJ, Weiner JL, Valenzuela CF. "Alcohol potently inhibits the kainate receptor-dependent excitatory drive of hippocampal interneurons." Proc Natl Acad Sci U S A. 2003;100:6813-8.
- Chiou LC, Chuang KC, Fan SH, How CH, Chen JK. "Does ethanol activate G-protein coupled inwardly rectifying K+ channels?" Neuroreport. 2002;13:163-5.
- Covernton PJ, Connolly JG. "Differential modulation of rat neuronal nicotinic receptor subtypes by acute application of ethanol." Br J Pharmacol. 1997;122:1661-8.
- Criswell HE, Ming Z, Griffith BL, Breese GR. "Comparison of effect of ethanol on N-methyl-D-aspartate- and GABA-gated currents from acutely dissociated neurons: absence of regional differences in sensitivity to ethanol." J Pharmacol Exp Ther. 2003;304:192-9.
- Dildy-Mayfield JE, Harris RA. "Ethanol inhibits kainate responses of glutamate receptors expressed in Xenopus oocytes: role of calcium and protein kinase C." J Neurosci. 1995;15:3162-71.
- Dopico AM, Anantharam V, Treistman SN. "Ethanol increases the activity of Ca(++)-dependent K+ (mslo) channels: functional interaction with cytosolic Ca++." J Pharmacol Exp Ther. 1998;284:258-68.
- Frye GD, Fincher A. "Sustained ethanol inhibition of native AMPA receptors on medial septum/diagonal band (MS/DB) neurons." Br J Pharmacol. 2000;129:87-94.
- Frye GD, Fincher AS, Grover CA, Griffith WH. "Interaction of ethanol and allosteric modulators with GABAA-activated currents in adult medial septum/diagonal band neurons." Brain Res. 1994;635:283-92.
- Jakab M, Weiger TM, Hermann A. "Ethanol activates maxi Ca2+-activated K+ channels of clonal pituitary (GH3) cells." J Membr Biol. 1997;157:237-45.
- Kobayashi T, Ikeda K, Kojima H, Niki H, Yano R, Yoshioka T, Kumanishi T. "Ethanol opens G-protein-activated inwardly rectifying K+ channels." Nat Neurosci. 1999;2:1091-7.
- Koski A, Ojanpera I, Vuori E. "Alcohol and benzodiazepines in fatal poisonings." Alcohol Clin Exp Res. 2002;26:956-9.
- Lovinger DM, White G. "Ethanol potentiation of 5-hydroxytryptamine3 receptor-mediated ion current in neuroblastoma cells and isolated adult mammalian neurons." Mol Pharmacol. 1991;40:263-70.
- Lovinger DM, White G, Weight FF. "Ethanol inhibits NMDA-activated ion current in hippocampal neurons." Science. 1989;243:1721-4.
- Mascia MP, Mihic SJ, Valenzuela CF, Schofield PR, Harris RA. "A single amino acid determines differences in ethanol actions on strychnine-sensitive glycine receptors." Mol Pharmacol. 1996;50:402-6.
- Ming Z, Knapp DJ, Mueller RA, Breese GR, Criswell HE. "Differential modulation of GABA- and NMDA-gated currents by ethanol and isoflurane in cultured rat cerebral cortical neurons." Brain Res. 2001;920:117-24.
- Moykkynen T, Korpi ER, Lovinger DM. "Ethanol inhibits alpha-amino-3-hydyroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor function in central nervous system neurons by stabilizing desensitization." J Pharmacol Exp Ther. 2003;306:546-55.
- Mullikin-Kilpatrick D, Treistman SN. "Inhibition of dihydropyridine-sensitive Ca++ channels by ethanol in undifferentiated and nerve growth factor-treated PC12 cells: interaction with the inactivated state." J Pharmacol Exp Ther. 1995;272:489-97.
- Oz M, Jackson SN, Woods AS, Morales M, Zhang L. "Additive Effects of Endogenous Cannabinoid Anandamide and Ethanol on alpha7-Nicotinic Acetylcholine Receptor-Mediated Responses in Xenopus Oocytes." J Pharmacol Exp Ther. 2005
- Rang HP, Dale MM, Ritter JM. Drug Dependence and Drug Abuse. In Pharmacology. Edinburgh: Churchill Livingstone. 2001. pp. 614-633.
- Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR. "Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons." Proc Natl Acad Sci USA. 2003;100:2053-8.
- Shiraishi M, Harris RA. "Effects of alcohols and anesthetics on recombinant voltage-gated Na+ channels." J Pharmacol Exp Ther. 2004;309:987-94.
- Solem M, McMahon T, Messing RO. "Protein kinase A regulates regulates inhibition of N- and P/Q-type calcium channels by ethanol in PC12 cells." J Pharmacol Exp Ther. 1997;282:1487-95.
- Sundstrom-Poromaa I, Smith DH, Gong QH, Sabado TN, Li X, Light A, Wiedmann M, Williams K, Smith SS. "Hormonally regulated alpha(4)beta(2)delta GABA(A) receptors are a target for alcohol." Nat Neurosci. 2002;5:721-2.
- Valenzuela CF, Bhave S, Hoffman P, Harris RA. "Acute effects of ethanol on pharmacologically isolated kainate receptors in cerebellar granule neurons: comparison with NMDA and AMPA receptors." J Neurochem. 1998;71:1777-80.
- Wallner M, Hanchar HJ, Olsen RW. "Ethanol enhances alpha 4 beta 3 delta and alpha 6 beta 3 delta gamma-aminobutyric acid type A receptors at low concentrations known to affect humans." Proc Natl Acad Sci USA. 2003;100:15218-23.
- Wan FJ, Berton F, Madamba SG, Francesconi W, Siggins GR. "Low ethanol concentrations enhance GABAergic inhibitory postsynaptic potentials in hippocampal pyramidal neurons only after block of GABAB receptors." Proc Natl Acad Sci U S A. 1996;93:5049-54.
- Wirkner K, Eberts C, Poelchen W, Allgaier C, Illes P. "Mechanism of inhibition by ethanol of NMDA and AMPA receptor channel functions in cultured rat cortical neurons." Naunyn Schmiedebergs Arch Pharmacol. 2000;362:568-76.
- Wright JM, Peoples RW, Weight FF. "Single-channel and whole-cell analysis of ethanol inhibition of NMDA-activated currents in cultured mouse cortical and hippocampal neurons." Brain Res. 1996;738:249-56.
- Ye JH, Tao L, Ren J, Schaefer R, Krnjevic K, Liu PL, Schiller DA, McArdle JJ. "Ethanol potentiation of glycine-induced responses in dissociated neurons of rat ventral tegmental area." J Pharmacol Exp Ther. 2001;296:77-83.
- Yu D, Zhang L, Eisele JL, Bertrand D, Changeux JP, Weight FF. "Ethanol inhibition of nicotinic acetylcholine type alpha 7 receptors involves the amino-terminal domain of the receptor." Mol Pharmacol. 1996;50:1010-6.