Abstract
Phenylacetic acids are efficiently synthesized from acetophenones via thiomorpholides under Phase Transfer Catalytic (PTC) condition. The reaction proceeds efficiently by using triethyl benzyl ammonium chloride (TEBA) as PTC and the reaction time decreased dramatically up to 1/5th to afford pure products in good to excellent yield.
Phenylacetic acids are very important compounds as they are showing broad range of biological activity, i.e., antibacterial2, analgesic3, virucidal4, prostaglandin synthetase response5, plant growth regulater6, etc. They are used as important intermediates in the syntheses of benzamido and phenylacetamido phenyl benzoxazole derivatives7, N-(aryl/hetero aryl acetyl) amino acid esters8, caprolactams9, benzodioxinones10, imidazoles11, N-(propynyl)aryl acetamides12, phenylfuranone13, halobenzyl isoquinolines14, estradiene and estratriene15, and anti cancer calcium channel blockers16.
Phenylacetic acids have been synthesized by (i) rhodium-catalyzed carbonylation17, (ii) hydrogenation of mandelic acid derivatives to the corresponding phenylacetic acid derivatives catalyzed by Pd/C18, (iii) carbonylation of benzyl chloride in the presence of the water-soluble complex PdCl2[PPh2(m-C6H4SO3Na)]2, and surfactants under two-phase conditions19, (iv) microwave induced Willgerodt-Kindler reactions of styrenes20, (v) microwave-assisted rapid hydrolysis and preparation of thioamides by Willgerodt-Kindler reaction21, (vi) production of phenylacetic acid by liquid phase oxidation of acetophenone with sulphur in the presence of aqueous ammonia22.
Although, all these methods have some limitations such as long reaction times, hazardous reaction conditions and use of microwave ovens which are not commonly available in all the research laboratories. Therefore in view of the above said limitations and their pharmaceutical and biological importance, there is an intense demand to develop a new and efficient method for the synthesis of biologically active phenylacetic acids under mild and ecofriendly reaction conditions. In this paper, we wish to report an efficient synthesis of phenylacetic acid and its derivatives in good to excellent yield under Phase Transfer Catalytic (PTC) conditions.
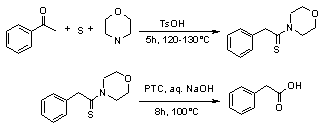
The reaction has been carried out at 100°C taking triethyl benzyl ammonium chloride (TEBA) as phase transfer catalyst. When thiomorpholides have been hydrolyzed with TEBA (114 mg, 0.05 mmol) under basic conditions the hydrolysis takes place within 8 h as indicated by TLC to afford phenylacetic acids in 80% yield. These results encouraged us to investigate the effect of TEBA (PTC) in hydrolysis, accordingly a series of thiomorpholides have been hydrolyzed into the corresponding phenylacetic acids in excellent yield.
It is noteworthy that the reaction time decreased dramatically almost up to 1/5th (i.e., from 24 to 5 h). Furthermore the products are isolated in almost pure form and do not require any further purification.
It is observed that the reaction follows the effect of substituent on benzene ring. The activated thiomorpholides are hydrolyzed faster than deactivated thiomorpholides. In the reaction the other sensitive groups such as chloro, bromo, methoxy, amino, hydroxy, alkyl, etc., are unaffected. All the products are characterized by comparing their physical data, 1H-NMR, MS and IR of authentic samples.
Thus, the present method is an efficient and facile for the synthesis of phenylacetic acids from acetophenones via their thiomorpholides under phase transfer conditions. The reaction is far superior to other earlier reported methods with regards to its simplicity, selectivity, reaction time, and yield (Table 1).
Table 1
PTC Willgerodt-Kindler reaction of acetophenones to phenylacetic acids
| Entry | Substrate A | Product B | Time | Yield |
| 1 | Acetophenone | Phenylacetic acid | 5h |
80% |
| 2 | 2-Acetylnaphthalene | 2-Naphthaleneacetic acid | 5h |
80% |
| 3 | 4-Isobutylacetophenone | 4-Isobutyl-phenylacetic acid | 6h |
78% |
| 4 | 4-Chloroacetophenone | 4-Chloro-phenylacetic acid | 8h |
78% |
| 5 | 4-Bromoacetophenone | 4-Bromo-phenylacetic acid | 8h |
78% |
| 6 | 2-Aminoacetophenone | 2-Amino-phenylacetic acid | 6h |
70% |
| 7 | 4-Hydroxyacetophenone | 4-Hydroxy-phenylacetic acid | 6h |
65% |
| 8 | 4-Methoxy-2-hydroxyacetophenone | 4-Methoxy-2-hydroxy-phenylacetic acid | 6h |
63% |
| 9 | 1-Hydroxy-2-acetylnaphthalene | 1-Hydroxy-2-naphthaleneacetic acid | 6h |
63% |
| 10 | 4-Methoxy-acetophenone | 4-Metoxy-phenylacetic acid | 6h |
60% |
Experimental
Typical procedure for the preparation of phenylacetic acid
Acetophenone (1.20 g, 10 mmol), sulfur (0.64 g, 20 mmol), morpholine (3 mL, 30 mmol), p-toluene sulfonic acid (0.06 g, 0.35 mmol) were added and held at reflux under constant stirring in an oil bath at 120–130°C for 8 h. After completion of the reaction as indicated by TLC, the reaction mixture was allowed to cool and 20% NaOH and triethyl benzyl ammonium chloride (TEBA) (114 mg, 0.05 mmol) were added to the reaction mixture and continued hydrolysis for further 8 h at 100°C. After completion of the reaction as indicated by TLC, the reaction mixture was cooled and filtered, the filtrate was acidified with HCl to pH 6 and then filtered off. The filtrate was further acidified to pH 2 and thus crude phenylacetic acid was obtained. The acid was then taken into 10% NaHCO3 solution and was washed with ethyl acetate (3x30 mL), separated the organic layer, and the aqueous layer was acidified with dilute HCl, to yield the pure phenylacetic acid as solid. In case of products from hydroxy acetophenones, the products were extracted into ethyl acetate. The dried organic layer was evaporated under reduced pressure to yield pure phenylacetic acid.