Abstract
Optically active 4-methoxy-α-methylphenylethylamine (1), a useful chiral building block in medicinal chemistry, was synthesized from L- or D-tyrosine by using a simple and efficient procedure via one-pot zinc reduction of the corresponding O-tosylate (4) in the presence of H2O and NaI in pure form.
Optically active α–methylphenylethylamines, such as 4-methoxy-α-methylphenylethylamine (1), are important chiral building blocks in medicinal chemistry. For example, one of our research targets is TA-20051, (R,R)-8-hydroxy-5-[1-hydroxy-2-[N-2-(4-methoxyphenyl)-1-methylethyl]aminoethyl]quinolin-2(1H)-one hydrochloride, which proved to be a potent, long-acting, and β2-selective agonist. The pure R form of amine 1 in large enough quantities is required for the bulk preparation of TA-2005 (R,R-isomer). Synthesis of the enantiomers of 1 via fractional crystallization of the tartrate2 of (±)-1 or via asymmetric reduction3 of the corresponding imine derivatives of 4-methoxyphenylacetone and (+)- and (-)-α-methylbenzylamines has been described previously. These methods have a number of disadvantages that include low resolution yield (9%) for the former reported method and loss of the chiral amine source in the hydrogenolysis step for the latter method. We therefore attempted to develop a new synthetic approach to both enantiomeric forms of 1.
Considering the structural similarity between commercially available L-tyrosine and R-1, the conversion of the CO2H group of tyrosine into a methyl group4 was investigated (Scheme 1).
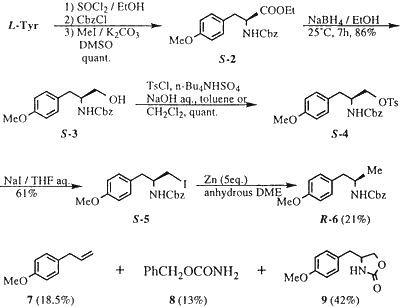
Esterification of L-tyrosine with EtOH in the presence of SOCl2 followed by N-protection with benzyloxycarbonyl chloride and O-methylation of the phenolic hydroxy group (CH3I/K2CO3 in DMSO) afforded S-2 in quantitative yield. Reduction of the ester group of S-2 with NaBH4 according to the method of Yamada et al.5 gave the corresponding alcohol (S-3) in 86% yield. Tosylation of S-3 was effectively performed by using n-Bu4N·HSO46 as a phase transfer catalyst in benzene / aqueous NaOH or in CH2Cl2 / aqueous NaOH to give the O-tosylate (S-4) in quantitative yield. Treatment of S-4 with NaI in refluxing aqueous THF afforded the corresponding iodide (S-5) in 69% yield.
Zinc reduction of the iodide (S-5) in anhydrous dimethoxyethane according to the method reported by Fujimoto et al.7 gave a mixture of products, which were separated by column chromatography (SiO2) providing the methyl derivative R-6 (21%) along with 7 (18.5%), 8 (13%), and 9 (42%), respectively (Scheme 2). Mechanistically, we believe that the addition of H2O to the reaction mixture is necessary to hydrolyze the corresponding RZnI. P. Kocovsky et. al8 reported the zinc reduction of steroidal alcohol via their mesylates and tosylates with NaI in refluxing DME, in the presence of H2O without any comment in their experimental section. Fujimoto et. al7 didn't mention the necessity of H2O in this reaction.
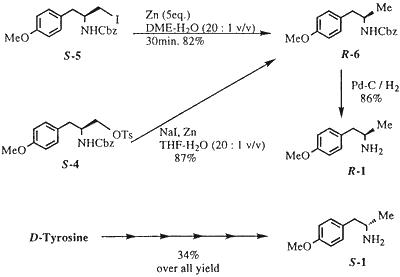
Zinc reduction of the iodide (S-5) was effectively carried out in aqueous dimethoxyethane to give the desired methyl derivative (S-6) in 82% yield We successfully utilized the above sequence in a one-pot conversion of S-4 into R-6. Thus, zinc reduction of S-4 in aqueous THF in the presence of NaI gave R-6 in 87% yield from S-4 (Scheme 3). Catalytic hydrogenolysis of R-6 on 10% Pd/C furnished R-1 in 86% yield (mp and [α]D, value were identical with the literature value).3 The optical purity of R-1 was determined to be >99.8% ee by HPLC analysis (see the Experimental Section). The corresponding enantiomer, S-1 was also obtained from D- tyrosine by using this method (Scheme 3). In this report, we have demonstrated that chiral 4-methoxy-α-methylphenylethylamine (1) can be prepared from tyrosine with high efficiency using very simple and robust chemistry which avoids the problems associated with previously reported approaches. This simple procedure for preparation of chiral a-methylphenylethylamines is also applicable to other our targets, such as TXA2 antagonists.
Experimental
All melting points were uncorrected. Column chromatography were carried out on silica Gel 60 (70-230 mesh ASTM) (Merck).
Synthesis of the R-amine (1) from L-Tyrosine.
(S)-Ethyl α-N-Benzyloxycarbonylamino-4-methoxybenzenepropionate, (S)-2.
To a stirred suspension of L-tyrosine (300 g, 1.657 mol) in EtOH was added dropwise SOCl2 (320 ml) within a period of 1 hr, under ice-salt cooling. After stirring at room temperature overnight, the reaction mixture was concentrated in vacuo, and treated with Et2O. The resulting precipitates were collected by filtration and washed well with Et2O to give pale yellow crystals (416 g, quant.). The hydrochloride salt was dissolved in a stirred mixture of AcOEt (2 L), K2CO3 (460 g), and H2O (1.4 L) and then benzyloxycarbonyl chloride (282 g) was added dropwise under stirring and ice-water cooling within a period of 0.5 hr. After stirring at 3-8°C for 0.5 hr, the AcOEt layer was separated, and successively washed with H2O, dil. HCl and brine, and dried over anhyd. Na2SO4. After removal of the solvent, the residue was dissolved in DMSO (2.6 L), and powdered K2CO3 (685 g) and CH3I (423 g) were added to this solution. The whole was stirred at room temperature for 16 hr. The reaction mixture was diluted with H2O, and extracted with benzene. The benzene extract was washed with aq. NaOH and brine, and dried over anhyd. Na2SO4. The solvent was concentrated in vacuo to give S-2 (605 g, quant.) as a pale yellow oil.
(S)-N-β-Benzyloxycarbonylamino-4-methoxyhenzenepropanol, (S)-3.
A solution of (S)-2 (605 g) in EtOH (4 L) was added to a stirred solution of NaBH4 (188 g, 4.95 mol) in EtOH (2 L) under ice-water cooling. After stirring at room temperature for 7 hr, excess NaBH4 was decomposed with slow addition of acetone (1.5 L) under ice-water cooling. The reaction mixture was neutralized with conc. HCl (412 ml). Insoluble material was filtered off, and the filtrate was concentrated in vacuo. The residue was extracted with AcOEt and the organic layer was successively washed with H2O, dil. aq NaOH, and brine, and dried over anhyd. Na2SO4. After removal of the solvent, the residue was crystallized from AcOEt-n-hexane to give (S)-3 (447 g, 86%) as a colorless solid, mp 93-99°C, [α]D20 -39.5° (c=1, MeOH). Recrystallization from AcOEt-n-hexane gave colorless scales mp 97-99°C.
(S)-N-Benzyloxycarbonyl-4-methoxy-α-p-tosyloxy methyl benzeneethanamine, (S)-4.
To a stirred mixture of (S)-3 (220.5 g, 0.7 mol), 10% aq. NaOH (1.12 L), p-tosyl chloride (143 g, 0.75 mol) and benzene (1.68 L) was added slowly a solution of n-Bu4N·HSO4 (47 g, 138 mmol) in H2O (200 ml) under ice-water cooling within a period of 15 min. The mixture was stirred at 15-20°C for 0.5hr. The benzene layer was separated, washed with H2O, and dried over anhyd. Na2SO4. The solvent was evaporated in vacua to leave (S)-4 (341 g, quant.) as a colorless solid, which was recrystallized from AcOEt-n-hexane to give colorless needles, mp 80-81°C.
(S)-N-Benzyloxycarbonyl-α-iodomethyl-4-methoxybenzeneethanamine, (S)-5.
A mixture of S-4 (34 g, 72.5 mmol), NaI (54 g), THF (500 ml), and H2O (25 ml) was refluxed for 2.5hr. After removal of the solvent, the residue was diluted with H2O, and extracted with benzene. The benzene extract was washed with dil. NaOH, and H2O, and then dried over anhyd. Na2SO4. After removal of the solvent, the residue was recrystallized from AcOEt-n-hexane to give (S)-5 (18.8 g, 61%) as colorless needles, mp 76-78°C. Recrystallization from AcOEt-n-hexane gave colorless needles, mp 82-84°C.
Preparation of (R)-N-Benzyloxycarbonyl-4-methoxy-α-methylbenzeneethanamine, (R)-6.
- Zinc reduction of (S)-5 in aqueous DME.
A stirred mixture of (S)-5 (21.3 g, 50 mmol), Zn powder (16.4 g, 250 mg-atom), DME (200 ml) and H2O (20 ml) was refluxed for 0.5hr. Inorganic material was removed by filtration and washed with EtOH. After removal of the solvent, the reside was treated with H2O and extracted with AcOEt. This last was washed with dil. HCl and H2O, dried over anhyd. Na2SO4 and evaporated in vacuo. The residue was purified by column chromatography on SiO2 with AcOEt-n-hexane (1:6 v/v) as an eluant to give R-6 (12.2 g, 81.6%) as a colorless solid. Recrystallization from AcOEt-n-hexane gave colorless pillars, mp 88-89°C.
- One-pot zinc reduction of (S)-4 in the presence of NaI in aqueous THF.
A stirred mixture of (S)-4 (1.41 g, 3 mmol), NaI (2.25 g, 15 mmol), Zn powder (2.0 g, 30 mg-atom), THF (20 ml), and H2O (1 ml) was refluxed for 2hr. The reaction mixture was treated in a similar manner as described in method 1 to give R-6 (780 mg, 87%) as a colorless solid, mp 87.5-88°C. This material was identical with a sample obtained by method 1 (IR, MS, and NMR spectra).
- Zinc reduction of (S)-5 in dry DME.
A stirred mixture of (S)-5 (2.13 g, 5 mmol), Zn powder (1.64 g, 25 mg-atom) and dry DME (20 ml) was refluxed for 5.5hr. After usual work-up described in method 1, the crude mixture was separated by column chromatography with n-hexane, then followed by AcOEt-n-hexane (1:5-1:1, v/v) as eluant to give the following compounds. 7 (137 mg, 18.5 %) as a colorless oil. R-6 (315mg, 21%) as a colorless solid, mp 87-88°C. This material was identical with a sample obtained method 1 (IR and NMR spectra). 8 (99 mg, 13%) as colorless needles (from AcOEt-n-hexane), mp 87-88°C.
(R)-4-Methoxy-α-methylbenzeneethanamine Hydrochloride, (R)-1.HCl
A mixture of R-6 (10.0 g, 33.4 mmol), THF (180 ml), and 10% aq. H2O (20 ml) was hydrogenated on 10% Pd-C (2.0 g) at atmospheric pressure and room temperature for 3hr. After removal of the catalyst, the filtrate was concentrated in vacuo and the residue was crystallized from EtOH-Et2O to give R-1.HCl (5.8 g, 86%) as colorless needles, mp 251-253°C (dec.), [α]D25 -23.2° (c=2.03, H2O), (mp 251-253°C (dec.). The optical purity of 1 was determined to be >99.8% ee by HPLC analysis.
Preparation of (S)-1 from D-Tyrosine.
(R)-Ethyl α-N-Benzyloxycarbonylamino-4-methoxybenzenepropionate, (R)-2.
D-Tyrosine (100 g, 0.55 mol) was treated as described above for the preparation of (S)-2 to afford (R)-2 (173g, 88.2%) as a colorless oil. This material was identical with (S)-2 (IR, MS, and NMR spectra).
(R)-N-α-Benzyloxycarbonylamino-4-methoxybenzenepropanol, (R)-3.
(R)-2 (173g, 0.485 mol) was reduced with NaBH4 (55.0 g, 1.45 mol) as described above for the preparation of (S)-3 to afford (R)-3 (98g , 65%) as colorless crystals (from AcOEt-iPr2O-n-hexane), mp 94-98°C. This material was identical with (S)-3 (MS and NMR spectra).
(R)-N-Benzyloxycarhonyl-4-methoxy-α-p-tosyloxy methylbenzeneethanamine, (R)-4.
(R)-3 (78.8 g, 0.25 mol) was treated as described above for the preparation of (S)-4 to afford R-4 (101.5 g 86%) as a colorless solid, which was recrystallized from AcOEt-iPr2O-n-hexane to give an analytical specimen as colorless needles, mp 79-82°C. This material was identical with (S)-4 (IR, Mass, and NMR spectra).
(S)-N-Benzyloxycarbonyl-4-methoxy-α-methylbenzeneethanamine, (S)-6.
(R)-4 (98.6 g, 0.21 mol) was converted to the methyl derivative (S)-6 as described above for the preparation of (R)-6 by method 2 (one-pot procedure) to give (S)-6 (53.7 g, 85%) as a colorless solid, which was recrystallized from AcOEt-n-hexane to give colorless pillars, mp 85-87°C. This material was identical with (R)-6.
(S)-4-Methoxy-α-methylbenzeneethanamine Hydrochloride, (S)-1.HCl
(S)-6 (55.1 g) was hydrogenated as described above for the preparation of (R)-1 to afford (S)-1·HCl (29.8 g, 80.3%) as colorless needles (from EtOH-Et2O), mp 251-253°C (dec.). This material was identical with R-1·HCl.