Abstract
Sufentanil, a potent anilidopiperidine analgesic, was synthesized from a simple thio-phenylethylamine via a six step sequence. The key parts of this synthesis involved an efficient construction of thiophenylethylpiperidone by aminomethano desilylation-cyclization followed by Swern oxidation and a direct regioselective N-nucleophilic spiral epoxide cleavage with aniline promoted by Lewis acids.
Introduction
Sufentanil (1), a well-known analgesic characterized by high potency, a rapid onset, and short duration of action, belongs to a series of the 4-propionanilidopiperidines which represents a class of morphine-like analgesics4. The extensive synthetic studies focused on 4-anilidopiperidine and its structural analogues1 have recently been carried out, although few synthetic studies on sufentanil have been reported. More recently, a series of work on the syntheses of sufentanil and its structural analogues have been carried out in our laboratory in conjunction with development of the novel analgesic agents. We have also reported an ideal synthetic route to N-arylpiperidines as a part of our successful results8 and we herein describe a total synthesis of sufentanil in full detail.
Experimental
Unless noted otherwise, all starting materials were obtained from commercial suppliers and were used without further purification. Tetrahydrofuran was distilled from sodium benzophenone ketyl. N,N-dimethylformamide and dimethyl sulfoxide were distilled under reduced pressure from calcium hydride and stored over 4A molecular sieves under argon. Dichloromethane, triethylamine, benzene, toluene, and pyridine were freshly distilled from calcium hydride. Nitromethane was distilled and stored over calcium hydride under argon. All solvents used for routine isolation of products and chromatography were reagent grade and distilled. Reaction flasks were oven dried at 120°C. Air and moisture sensitive reactions were performed under an argon atmosphere. Flash column chromatography was performed using silica gel 60 (230-400 mesh, Merck) with indicated solvents. Thin-layer chromatography was performed using 0.25 mm silica gel plates (Merck). Melting points were measured on a Buchi melting point apparatus and were uncorrected.
4-Hydroxy-1-(2-thiophenethyl)-piperidine (7)
A heterogenous mixture of allyltrimethylsilane (2.0 mL, 12.6 mmol), water (2.8 ml), thiophenylethylammonium trifluoroacetate (2.02 g, 8.4 mmol) and 37% aqueous formaldehyde (1.87 ml, 23.1 mmol) in water was stirred at 58°C for 24 hours. Water was added and then the reaction mixture was alkalized with 1N NaOH and extracted with CH2Cl2. The organic extracts were washed with brine, dried over anhydrous MgSO4 and evaporated. The residue was purified by column chromatograph) (MeOH/CHCl3, 1:10) to give the piperidine 7 (0.77 g, 43.3%) as a white solid.
1-(2-Thiophenethyl)-4-piperidone (8)
Oxalyl chloride (0.44 ml, 5.17 mmol) dissolved in CH2Cl2 (12 ml) was placed in a flask under nitrogen. The flask was cooled to -78°C and DMSO (0.73 mL) in CH2Cl2 (2.15 mL) was added dropwise for about 5 min. Stirring was continued at -78°C for an additional 10 min followed by addition of the alcohol 7 (575 mg, 2.72 mmol) in CH2Cl2 (2 ml) for about 5 min. After additional 15 min with stirring, triethylamine (2.88 mL, 20.7 mmol) was added for about 5 min with stirring and then the reaction mixture was allowed to warm to room temperature. Water (12 ml) was added and the aqueous layer was reextracted with CH2Cl2 (50 mL). The combined organic layers were washed with brine, and dried over anhydrous MgSO4. The filtered solution was concentrated and the residue was purified by column chromatography to give the ketone 8 (556 mg, 98%) as a white solid.
4-Anilinomethyl-4-hydroxy-1-(2-thiophenethyl)piperidine (10)
and 2-Anilino-1-(2-thiophenethyl)-4-piperidylmethanol (11)
Aniline (0.16 ml, 1.76 mmol) and triethyloxonium tetrafluoroborate (0.44 ml of 1M solution in CH2Cl2, 0.44 mmol) was added to the oxirane 9 (196 mg, 0.88 mmol) in CH2Cl2 (8 ml) at -78°C. The mixture was stirred at -78°C for 6 h. After quenching with water, the solution was basified with 1 N NaOH and extracted with CH2Cl2 (20 ml). The organic layers were dried over anhydrous MgSO4 and evaporated. The residue was purified by column chromatography to give piperidylmethanol 11 (19.7 mg, 6.2%), and hydroxypiperidine 10 (15.5 mg, 4.8%).
N-[4-Methoxymethyl-1-(2-thiophenethyl)]-4-piperidyl-N-phenylpropanamide (Sufentanil) (1)
A mixture of piperidylamine 12 (1.4 mg, 0.004 mmol) and propionic anhydride (1.2 mL, 0.009 mol) was refluxed with stirring for 19 hours. Cooled to 0°C, the mixture was basified with NH4OH and extracted with CH2Cl2. The organic layers were dried over MgSO4 and evaporated. The residue was chromatographed to give sufentanil 1 (0.8 mg, 50%).
Results And Discussion
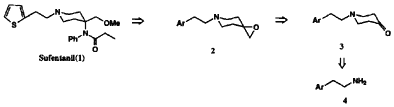
Our synthetic approach shown in Scheme 1 involved an efficient construction of the piperidone skeleton via sequential aminomethano desilylation-cyclization and oxidation of the resulting hydroxypiperidine. The N-arylalkylpiperidone 3 was also directly converted to the spiroepoxypiperidine 2 as a second potential intermediate which reacted with aniline to afford 4-anilinopiperidine along with 4-anilinomethylpiperidine as its regioisomer.

The intramolecular Mannich type cyclization as a key step of our synthesis was conducted, as outlined in Scheme 2, by a reaction of trifluoroacetic acid salt of the starting thiophenethylamine 5 with 1.1 equivalent of allyltrimethylsilane and 2.3 equivalent of 37% aqueous formaldehyde3,5. The resulting 4-hydroxythiophenethylpiperidine 7 was oxidized to piperidone 8 by the introduction of epoxide moiety. It should be noted that only Swern oxidation was effective for oxidation of hydroxypiperidine7.
The one step conversion of thiophenylethylpiperidone 8 to the spiroexpoxide 9 as a second potential intermediate was achieved by dimethylsulfonium ylide treatment as shown in Scheme 2. The variety of reaction conditions for regioselective ring opening of the epoxide at more substituted carbon by aniline were examined.
Triethyloxonium tetrafluoroborate (Et3O+BF4-) as Lewis acid in methylene chloride below -78°C turned out to be the best choice for the highest regioselectivity (1.8:1) in favor of 11. Usage of other Lewis acids afforded the regioisomer 10 as a predominant product or only byproducts by retro-Mannich type reaction. Although the regioselectivity and yields are not satisfactory yet, the direct introduction of aniline nucleophile to spiroepoxide at more substituted carbon enables the two step conversion of the arylalkylpiperidone to the highly advanced sufentanyl intermediate. Methylation of piperidylmethanol (11) was achieved by an initial reaction with diazomethane6. Finally, the synthesis was completed by the known acylation of anilinopiperidine 12 with propionic anhydride2 to afford the desired product which was identical in all aspects to the authentic sufentanil.
In conclusion, the total synthesis of sufentanil was accomplished by only six step reaction sequence. The key features of this synthesis include the efficient construction of thiophenethylpiperidine skeleton from thiophenethylamine and the regioselective ring opening of spiroepoxide at more substituted carbon by N-nucleophile. This methodology is applicable to the efficient syntheses of other anilinopiperidine analgesics and their structural analogues.