Abstract
The syntheses and physical properties are described for 2,5-dimethoxy-4-methylthio- phenylethylamine and 2,5-dimethoxy-4-methylthiophenylisopropylamine. The latter compound is the sulfur analog of the psychotomimetic phenylisopropylamines 2,4,5-trimethoxyphenylisopropylamine and 2,5-dimethoxy-4-methylphenylisopropylamine wherein the methylthio group replaces a methoxy group or a methyl group, respectively. This compound is predicted to be about 30 times as active as mescaline.
The 2,4,5-substituent orientation provides maximum potency in the trisubstituted psychotomimetic phenylisopropylamines1, and recent studies focused attention upon the pharmacological importance of the chemical nature of specific 4-substituents2,3. 2,5-Dimethoxy-4-methylphenylisopropylamine (I) is a potent compound which produces severe sensory disturbances and hallucinations. Increasing the length of the alkyl chain at the 4-position results in an increase in activity. Optimum activity is realized when the alkyl chain is three carbons2,3, and this result may be related to lipid partitioning4.
The qualitative nature of intoxication induced also varies with the 4-substituent. For example, 2,5-dimethoxy-4-ethylphenylisopropylamine (II) and 2,5-dimethoxy-4-bromophenylisopropylamine (III) are about 2-10 times more potent, respectively, than I5-7 in producing threshold central nervous system effects. In contrast, neither elicits the severe visual disturbances characteristic of I. Both the quantitative and the qualitative nature of the drug's action apparently are determined by the 4-substituent, although these relationships have not been defined clearly.
Discussion
One route for the metabolism of I is through oxidation of the 4-methyl group8,9. It might be anticipated that such oxidation would be less favored in the higher alkyl homologs and that it could not occur with the 4-bromo compound. It was shown recently that II is metabolized in rats at the 4-ethyl position, but more slowly than with I10, and that III generates no detectable inorganic bromide in humans11.
The ability of a compound to produce hallucinatory disturbances may be lost when the 4-substituent is resistant to oxidation. The concept that facile oxidation of the 4-function might be involved in the production of hallucinations by psychotomimetic amphetamine derivatives may receive support through study of an amphetamine derivative with a 4-substituent that is unusually susceptible to oxidative metabolism.
The sulfur atom would appear to be an excellent choice for such a substituent. Metabolically, it should retain the chemical properties of oxygen in reactions such as S-demethylation but should allow the formation of sulfoxides and sulfones as products of oxidative metabolism12. The lipophilicity, electronic character, and size of sulfur also seem ideal. A comparison of the σp and π values for methoxy, methyl, methylthio, and bromo shows that the methylthio group lies intermediate between methyl and bromo13-15. Barfknecht et al.4 correlated human activity with octanol-water partition coefficients; by using the principles of additivity16, one can predict that the methylthio analog should be some 30 times as potent as mescaline.
This paper describes the syntheses and properties of 2,5-dimethoxy-4-methylthiophenylisopropylamine (IV) and 2,5-dimethoxy-4-methylthiophenylethylamine (V). Compounds IV and V, as well as the intermediates IX, Xa, and Xb, have not been reported previously.
Scheme I
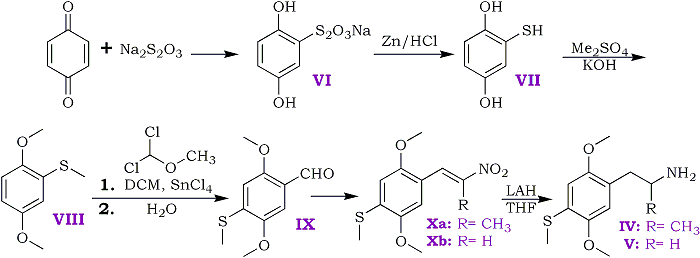
Experimental
2,5-Dihydroxyphenylthiosulfuric Acid, Sodium Salt (VI)
The method of Alcalay17 was modified as follows (Scheme I). A solution of 43.2 g of benzoquinone (0.4 mole) in 200 ml of acetic acid was added over 10 min to a solution of 149 g of sodium thiosulfate in 300 ml of water. The mixture was stirred for 1 hr and then reduced under vacuum to a thick syrup. The residue crystallized on standing and was slurried in a saturated salt solution and filtered. The solids were washed with small portions of a saturated salt solution, sucked dry, and finally dissolved in methanol and filtered through diatomaceous earth [Celite]. The methanol filtrate was concentrated in vacuo, yielding 67g (68%) of VI as a yellow powder. This powder was used directly in the next step.
2,5-Dihydroxythiophenol (VII)
Fifty grams of VI (0.205 mole) was dissolved in a mixture of 200 ml of water and 400 ml of concentrated hydrochloric acid. Zinc dust (250 g) was added slowly over 1.5 hr, with external ice bath cooling applied as needed to keep the temperature at 50-60?C. Adequate ventilation was needed because a large quantity of hydrogen sulfide was generated. Near the end of the reduction, the reaction mixture changed to a semi-solid gray mass. An additional 50 ml of concentrated hydrochloric acid was added to the mixture, and the solution was decanted from unreacted zinc metal. The aqueous acid solution was extracted with ether (6x100 ml), and the pooled extracts were washed with saturated sodium chloride solution (3x75 ml), dried (sodium sulfate), and concentrated in vacuo. The resulting yellow solid was recrystallized from ether to yield 24.7 g (85%) of pale-yellow needles, mp 118-119?C [lit.17 mp 118?C].
2,5-Dimethoxymethylthiophenol (VIII)
The method of Butenandt et al.18 was modified as follows. To 200 ml of 9 N KOH (1.8 M) stirred under nitrogen was added 24 g (0.169 mole) of VII. Methyl sulfate (160 g, 1.27 mole) was added, with vigorous stirring, over 2 hr. The temperature (50-60?C) was maintained by the rate of addition. The mixture was then heated to reflux for 3 hr, stirred overnight at room temperature, and filtered. The filtrate was extracted with 6x100 ml of ether, and the combined ether extracts were washed with 2x50 ml of saturated sodium chloride solution, dried (sodium sulfate), and concentrated in vacuo. The residue was vacuum distilled to give 25.9 g (83%) of product, bp 86-88?C/0.04 mmHg [lit.18 bp 96-97?C/0.07 mmHg] and mp 33-34?C [lit.18 mp 35?C].
2,5-Dimethoxy-4-methylthiobenzaldehyde (IX)
The aldehyde was prepared using the method of Rieche et al.19. A solution of 6.07g (0.033 mole) of VIII in 40 ml of dry dichloromethane under nitrogen was cooled in an ice bath. To the solution was added 13.02 g (0.05 mole) of stannic chloride over 2 min. Dichloromethyl methyl ether, 3.45 g (0.03 mole), was then added dropwise over 5 min, and stirring was continued with ice bath cooling for 15 min. The reaction was allowed to warm to room temperature over 30 min and was stirred for an additional 1 hr, at which time hydrogen chloride evolution had ceased. Then the mixture was slowly poured onto 15 g of ice in a separator, and the aqueous layer was separated and discarded. The organic phase was washed with 3x25 ml of 3 N HCl and 3x25 ml of saturated sodium chloride solution and dried (sodium sulfate), and the solvent was removed in vacuo. The solid residue was dissolved in methanol, filtered, and recrystallized from methanol-water to give 5.86 g (92%) of yellow needles. TLC (silica gel, chloroform) showed only one product. An analytical sample was further purified via the sodium bisulfite adduct and recrystallized from methanol-water, mp 99-100?C;
NMR (CDCl3):
δ 2.48 (s,3H,SCH3), 3.92, 3.97 (2s,6H,OCH3), 6.74, 7.29 (2s,2H,ArH), 10.45 (s,1H,CHO).
The ylidinemalononitrile derivative of IX was prepared from equal weights of the benzaldehyde and malononitrile in ethanol with triethylamine catalysis, mp 185-186?C after recrystallization from ethanol.
1-(2,5-Dimethoxy-4-methylthiophenyl)-2-nitropropene (Xa)
A solution of 2.3 g (0.011 mole) of IX in 7.5 ml of nitroethane containing 0.45 g of ammonium acetate was heated on the steam bath for 6 hr. Removal of most of the nitroethane in vacuo, followed by the addition of 10 ml of methanol, gave crystals. After filtering and washing with cold methanol, the product was recrystallized from 140 ml of boiling ethanol, yielding 1.8 g (62%) of bright-orange crystals, mp 137-138?C;
NMR (CDCl3):
δ 2.42 (s,3H,CH3), 2.50 (s,3H,SCH3), 3.88 (s,6H,OCH3), 8.29 (br.s,1H, =CH), 6.79, 6.83 (2s,2H,ArH).
1-(2,5-Dimethoxy-4-methylthiophenyl)-2-nitroethylene (Xb)
Essentially the same procedure as for Xa was used, but use of nitromethane instead of nitroethane gave the homologous nitrostyrene. Recrystallization from ethanol gave rust-orange crystals (yield 68%), mp 165.5-166?C.
2,5-Dimethoxy-4-methylthiophenylisopropylamine (IV)
To a gently refluxing suspension of 1.4 g of lithium aluminum hydride in 40 ml of tetrahydrofuran was added dropwise a saturated solution of 1.7 g (0.006 mole) of Xa in tetrahydrofuran. Reflux with stirring was continued for 7 hr. The reaction mixture was then cooled externally with ice water; 1.6 ml of water, 1.6 ml of 15% NaOH, and finally 4.8 ml of water were added dropwise. Stirring was continued until no gray-colored solids remained. The mixture was filtered, the filter cake was washed with tetrahydrofuran, and the mother liquor and washings were combined and evaporated to dryness in vacuo.
The free base was a white solid; an analytical sample, after recrystallization from hexane, had a melting point of 91-93?C. The product was dissolved in 25 ml of 2-propanol, titrated with concentrated hydrochloric acid to a persistent pink color (0.57 ml), and diluted with 100 ml of anhydrous ether. After a few moments, there was spontaneous formation of white crystals, 1.2 g (68% yield), mp 204-205?C.
2,5-Dimethoxy-4-methylthiophenylethylamine (V)
The same procedure as was used for IV but with Xb instead of Xa gave the corresponding phenethylamine hydrochloride (V) as white crystals from 2-propanol-ether (57% yield). Recrystallization from ethanol gave melting point of 240-241?C with prior darkening.