Synthesis of Phenylacetonitriles from BenzaldehydesRhodanine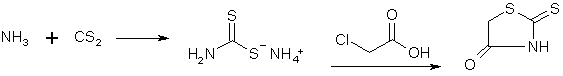
Dry ammonia gas is passed for three and one-half hours into a mixture of 250 g. of carbon disulfide, 200 cc. of alcohol and 200 cc. of ether, cooled well in a freezing mixture. The contents of the flask then represent an almost solid cake of pale yellow crystals of ammonium dithiocarbamate. These are filtered by suction, washed well with 50 cc. of cold alcohol, followed by 100 cc. of ether. The material must be used immediately; on standing it deteriorates rapidly. In the meantime a solution of sodium chloroacetate has been prepared by adding a solution of 98 g. of sodium hydroxide in 150 cc. of water to a solution of 238 g of chloroacetic acid (Eastman practical) in 200 cc. of water. Sodium carbonate is added until the solution is neutral to litmus. To the solution thus prepared the ammonium dithiocarbamate, which should weigh 280-310 g. is added gradually, under stirring and good cooling, the color turning dark at first, gradually lightening and finally becoming a straw yellow if all has gone well. This solution is then added under stirring to 300 cc. of concentrated hydrochloric acid which has been heated to 80-90°. The rhodanine slowly separates in glistening, pale yellow prisms. After an hour's standing the flask with its contents is cooled in an icebath, the rhodanine filtered and washed well with water; yield of air-dried material 210-230 g., m. p. 162°. This rhodanine is pure enough for practically all condensations, but may be purified by recrystallization from alcohol, mp 170°, almost colorless. Veratralrhodanine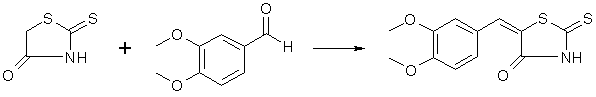
To a solution of 100 g. of veratrumaldehyde and 80 g of rhodanine in 400 ml of hot glacial acetic acid, 150 g. of fused sodium acetate is added and the whole boiled for one-half hour with occasional shaking. The veratralrhodanine soon separates in orange colored crystals. The whole mass is poured into 3 liters of water, the crystals filtered off, washed well with water, then with little alcohol and ether; yield 162g, 96% of the theoretical; mp 232°, recrystallized from acetone. Cleavage of Veratralrhodanine with Alkali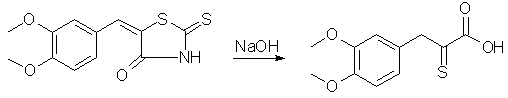
Forty-four grams of veratralrhodanine is suspended in 200 cc. of 15% sodium hydroxide and heated on a vigorously boiling water-bath with occasional shaking until all the material dissolves (about one-half hour). The alkaline solution is cooled well in a freezing mixture and the acid precipitated rapidly with 200 cc. of 10% hydrochloric acid. It is well not to add the acid too slowly, for the material which first precipitates goes back into solution and the yield is lowered. Slow addition necessitates use of more than the quantity of acid indicated above, and several runs have shown that this quantity is more than sufficient. If the precipitate which first comes out is allowed to go back into solution, additional acid precipitates a gum which crystallizes with difficulty and a considerable quantity of hydrogen sulfide is evolved. The amorphous acid which separates soon crystallizes on standing in the freezing mixture to a pale yellow powder; yield practically quantitative, m. p. 179° after recrystallization from methyl alcohol. For the best results it is not recommended to run larger quantities for the cleavage. Indeed smaller runs are to be preferred, especially since the process is simple and several runs can be made simultaneously. The succeeding operations go well with any quantities. alpha-Oximino-beta-3,4-dimethoxyphenylpyruvic Acid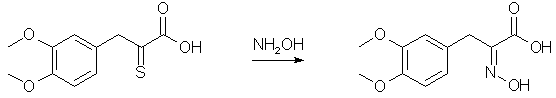
To a solution of sodium ethylate prepared from 22.5 g. of sodium and 650 cc. of ethyl alcohol is added a warm solution of 67.5 g. of hydroxylamine hydrochloride in 60 cc. of water. The solution of hydroxylamine is filtered from the precipitated sodium chloride and poured onto 75 g. of the thioketo acid once recrystallized from methyl alcohol. The resulting solution is heated on the water-bath for about twenty minutes and the alcohol removed under diminished pressure. The residual solid mass is dissolved in 160 cc. of 5% sodium hydroxide, filtered from sulfur, well cooled and cautiously acidified with 150 cc. of 10% hydrochloric acid (Caution! H2S evolution!). On scratching the oximino acid crystallizes in almost colorless flakes. It is filtered, washed with 200 cc. of water and dried in a vacuum desiccator over potassium hydroxide overnight; yield 72 g. Decomposition of alpha-Oximino-beta-3,4-dimethoxyphenylpyruvic Acid to 3,4-Dimethoxyphenyl-acetonitrile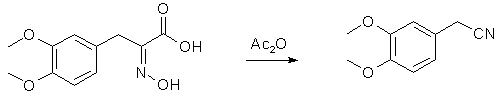
Ninety-three grams of crude dry oximino acid is suspended in 300 cc. of acetic anhydride and warmed cautiously. Effervescence begins on slightest warming and the oximino acid gradually goes into solution. After all decomposition is over, the acetic anhydride is removed under diminished pressure and the residue taken up in water and ether. The ethereal layer is washed well with sodium carbonate and distilled. At 187-188° an almost colorless liquid distils, yield 65 g. This represents almost pure nitrile and crystallizes on cooling, m. p. 64-65°. For the catalytic hydrogenation to homoveratrylamine it is recrystallized once from methyl alcohol. All these reactions have been carried out on piperonal and anisaldehyde with equally good yield. Reference: JACS 57, 1126-1128 (1935) |