|
Synthesis of diphenyl-2-pyrrolidinyl-methanol and diphenyl-2-pyrrolidinyl-methane author unknown[ Back to the Chemistry Archive ] Introduction:
(S)-diphenyl-2-pyrrolidinyl-methane is a CNS stimulant, chemically very similar to pipradrol (another CNS stimulant, It just has a pyrrolidine ring instead of a piperidine ring, and no hydroxy group. It is also somewhat similar to methylphenidate. According to the patent it is active in dosages from 25mg to 100mg. LD50 is 28mg/kg IV. More pharmacology data is in the patent[1]. In the book Designer Drugs Directory the intermediate Diphenyl-2-pyrrolidinyl-methanol is described as being active in 2-5mg. Interestingly the methanol is also used for sterospecific reduction of ketones to alcohols[2], so you could be able to get it through legal channels. The binding affinity values for Diphenyl-2-pyrrolidinemethanol at the cocaine site of the DAT are as follows [6].
The unit Ki is a measure of receptor binding affinity of a compound, its "potency" roughly. The lower the Ki, the higher the potency. Ki is a relative value assigned to the compound when compared to a standardized ligand for the receptor, and not a physical value. Kibinding is how well the compound binds to the DAT receptor. Kiuptake is how well the compound is at inhibiting the functioning of the DAT (simply binding to the receptor does not equal effective inhibition). Cocaine inhibits the DAT, thereby blocking DA reuptake and increasing the DA concentration in the synapse. The more DA the merrier. The authors of the patent wants to medicate cocaine addiction by making a compound with low Kibinding but high Kiuptake, i.e. a molecule that sits tight in the receptor, but without doing anything. If it binds tighter to the DAT than cocaine does, then it will competitively displace cocaine from the receptor if both are present simultaneously (making cocaine ingestion ineffective). This is similar to the way Naloxone and Naltrexone effects the opiate receptor. On the other hand, for a compound to have as much recreational potential as possible, we would like a compound which sits tight in the receptor and while there block the DAT as effectively as possible. Thus we want both Kibinding and Kiuptake to have low values. As a fundamental limitation to the effiacy of a compound consists of its ability to bind to its target, Kibinding is always higher, or at most equal to Kiuptake. Therefore the ideal recreational DAT ligand has a ratio of 1:1 between the values, as in that every time a molecule binds to a DAT receptor, it manages to shut it down. If the ratio is 1:2, the "success rate" is 50% (for Cocaine itself, the ratio is 1:1.67). Also, beside this ratio, we want the needed dosage of the drug to be as low as possible (for economic reasons, to name one thing), so therefore a low intrinsic Ki value is desirable. Glossary:
Synthesis:(S)-Diphenyl-2-pyrrolidinyl-methanol [1] 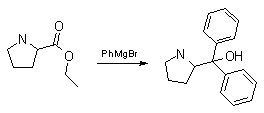 Phenylmagnesium bromide was prepared from 61g Mg and 372g bromobenzene in 2L ether. Then 0.7L anisol is added, and the major part of the ether is distilled off. Then a solution of 52.2g (S)-proline ethyl ester in 80ml anisol is added, under light reflux. After 30 minutes of reflux one pours the solution into a solution of 1000g crushed ice and 372ml concentrated hydrochloric acid , filters, dissolves the precipitate in hot water and makes the solution basic with ammonia in the presence of ether. The ether layer is decanted and evaporated dry. The residue is washed with ethanol and dried. Thus obtained is 20g of (S)-diphenyl-2-pyrrolidinyl-methanol, in the form of colorless crystals. (S)-Diphenyl-2-pyrrolidinyl-methane [1]  Into a solution of 125g (S)-diphenyl-2-pyrrolidinyl-methanol in 450ml acetic acid is added 1265ml of a solution of HI in 2N acetic acid. This is then heated to 115°C on the oil bath for 5 minutes and then cooled quickly. To the cooled mixture is added 1260ml of a saturated solution of sodium thiosulphate, and then evaporated to dryness. The residue is put into 630ml water and filtred. The precipitate is dissolved in 1000ml of methanol, and filted. The organic phase is made basic with soda. Then 600ml water is added, the methanol is removed by distillation, and the aquous solution is extraced with ether. The ether solution is evaporated to dryness. Thus obtained is 115g of crude (S)-diphenyl-2-pyrrolidinyl-methane. This is dissolved in dry ether and concentrated hydrochloric acid is added until PH = 4, and then filtered. The precipitate is dried and washed with acetone and ether. The hydrochloride is purified by recrysalisation from boiling water. One obtains 70 to 80g of the hydrochloride of (S)-diphenyl-2-pyrrolidinyl-methane. Synthesis of (S)-2-(Diphenylhydroxymethyl)pyrrolidine [3](S)-1-(Trimethylsilyl)pyrrolidinyl-2-trimethylsilylcarboxylate  11,5 g (100 mmol) (S)-Proline and 34,90 g (240 mmol) N,N-diethyl-trimethylsilylamine were heated in an oil bath at 110-120°C, and the formed diethylamine was continuously distilled off. After 6h the heating was terminated since no more diethylamine was formed. Vacuum distillation of the residue gave 24.53 g (94.5%) of (S)-1-(trimethylsilyl)pyrrolidinyl-2-trimethylsilylcarboxylate (bp 97°C/14 mbar) as a very hydrolysis-sensitive clear liquid. (S)-2-(Diphenylhydroxymethyl)pyrrolidine 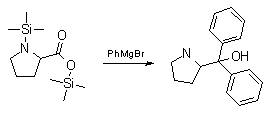 The Grignard complex prepared from 7.29 g (300 mmol) magnesium and 47,1 g (300 mmol) bromobenzene in 350 ml absolute Et2O was added dropwise to 19.46 g (75 mmol) of the above ester in 30 ml absolute Et2O at 0-20°C under argon atmosphere. The reaction solution was heated under reflux for 4h, cooled to 0°C and poured slowly with stirring into 450 ml 2N ice-cold hydrochloric acid (360 g ice + 90 ml conc. HCl). The white precipitate was filtered by suction, washed with methyl tert-Butylether, water and recrystallized from Methanol/methyl tert-Butylether. By collecting several fractions, there was obtained 14,26 g (49,2 mmol) of crude (S)-2-(Diphenylhydroxymethyl)pyrrolidine.HCl with a purity of 65.6%, mp 285-290°C. The crude (S)-2-(Diphenylhydroxymethyl)pyrrolidine.HCl was added to a mixture of 100 ml 1N NaOH, 5 ml triethylamine and 200 ml methyl tert-Butylether, and the mixture stirred until two clear phases formed (about 5h). The phases were separated and the aqueous phase extracted twice with methyl tert-Butylether. The pooled organic phases were washed twice with 20% aqueous NaCl solution, dried over MgSO4, filtered and the solvent removed under vacuum at a bath temp of 40°C. The residue was taken up in hot n-heptane, and upon cooling clear, yellowish crystals precipitated. Thus, there was obtained 12.01g (63%, 47.5 mmol) of (S)-2-(Diphenylhydroxymethyl)pyrrolidine (mp 76.5-78°C). DL-alpha-(2-Pyrrolidyl)benzhydrol Hydrochloride [4]dl-Proline ethyl ester (4.7g) was added dropwise at room temp to a stirred solution of phenylmagnesium bromide (4 molar equivalents) in ether. After addition of the ester, the mixture was boiled under reflux for 2h and then hydrolyzed with dilute ammonium chloride. The ether layer was separated and the aqueous solution extracted thrice with toluene. The combined extracts wre was washed with water, dried over sodium sulfate and evaporated in vacuo to yield an oil (4.5g). It was converted to the hydrochloride and recrystallized from methanol-ether, mp >250°C. An aliquot of the hydrochloride salt converted to the free base and recrystallized from ethanol-water had mp 81-82°C (lit. [5] mp 83°C). (S)-1,1-Diphenylprolinol [7]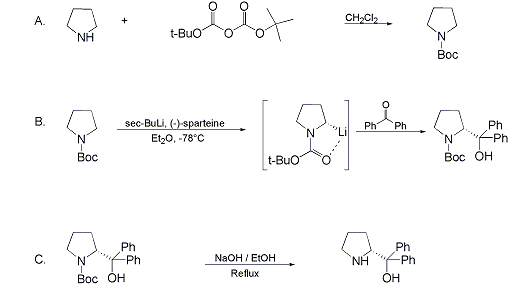 A. N-(tert-Butoxycarbonyl)pyrrolidine. A 500-mL round-bottomed flask, equipped with magnetic stirring bar, is charged with dichloromethane (CH2Cl2) (120 mL) and pyrrolidine (11.3 mL, 133 mmol) (Note 1). The flask is fitted with a 60-mL, pressure-equalizing addition funnel vented through a mineral oil bubbler and charged with a solution of di-tert-butyl dicarbonate (24.6 g, 112 mmol) in CH2Cl2 (35 mL) . After the pyrrolidine solution is cooled to 0°C in ice, the colorless dicarbonate solution is added dropwise over a period of 30 min, and the resulting solution is stirred at room temperature for 3 hr (Note 2). The solvents are then removed under reduced pressure, and two consecutive Kugelrohr distillations of the residual oil (oven temperature 80°C at 0.2 mm) afford 16.6 g (87%) of N-Boc-pyrrolidine as a colorless oil (Note 3). B. (R)-(+)-2-(Diphenylhydroxymethyl)-N-(tert-butoxycarbonyl)pyrrolidine. An oven-dried, 2-L, three-necked flask, equipped with a magnetic stirring bar and a thermocouple (Note 4), is charged with (-)-sparteine (30.2 mL, 131 mmol) (Note 5), N-Boc-pyrrolidine (15.0 g, 87.6 mmol), and anhydrous ether (900 mL) (Note 6). The solution is cooled to -70°C (dry ice/acetone bath) (Note 4). To this solution is added sec-butyllithium (96 mL, 1.16 M in cyclohexane, 111 mmol) (Note 7) and (Note 8) dropwise over a period of 35 min (Note 9). The reaction is then stirred at -70°C for 5.5 hr (Note 10). After this interval, a solution of benzophenone (25.5 g, 140 mmol) (Note 11) in anhydrous ether (200 mL) is added dropwise over a period of 1.25 hr (Note 9). The dark green to greenish-yellow suspension is maintained at -70°C for 2.0 hr, and the reaction is then quenched by dropwise addition of glacial acetic acid (8.5 mL, 150 mmol) over a period of 15 min. The resulting lemon-yellow suspension is allowed to warm slowly to room temperature over a period of 12 hr, during which time the mixture becomes cream colored. After the solution is warmed to 25°C, 5% phosphoric acid (H3PO4) (150 mL) is added to the reaction mixture, and the resulting biphasic mixture is stirred for 20 min. The layers are partitioned and the organic phase is washed with additional 5% H3PO4 (3 x 150 mL). Combined aqueous phases are extracted with ether (3 x 200 mL). The original organic phase and the ethereal extracts are combined, washed with brine (200 mL), dried over magnesium sulfate (MgSO4), filtered, and the solvents are removed under reduced pressure to afford crude product as an off-white solid. The crude (R)-(+)-2-(diphenylhydroxymethyl)-N-(tert-butoxycarbonyl)pyrrolidine is purified by recrystallization from a mixture of hexanes-ethyl acetate (675 mL, 20 : 1, v/v) affording in two crops 20.9-22.0 g (73?74%) of analytically pure product as a white solid (Note 12) having greater than 99.5% ee (Note 13). Sparteine is recovered by making the aqueous phases basic with aqueous 20% sodium hydroxide (NaOH) (160 mL) (Note 14). The aqueous phase is extracted with Et2O (4 x 150 mL), and the combined organic phases are dried over potassium carbonate (K2CO3), filtered, and the solvents removed under reduced pressure to afford 30.3 g (98%) of crude, recovered sparteine as a pale yellow oil (Note 15). Fractional distillation of the residual oil from calcium hydride (CaH2) (Note 5) affords 27.0 g of sparteine (88%) suitable for reuse. C. (R)-(+)-2-(Diphenylhydroxymethyl)pyrrolidine. A 1-L, round-bottomed flask, equipped with a magnetic stirring bar, is charged with 325 mL of absolute ethanol and NaOH (27.0 g, 675 mmol). The NaOH is dissolved with vigorous stirring, and (R)-(+)-2-(diphenylhydroxymethyl)-N-(tert-butoxycarbonyl)pyrrolidine (22.0 g, 62.3 mmol) is added (Note 16). The flask is fitted with a reflux condenser, and the resulting milky white suspension is heated to reflux for 2.5 hr. The suspension is cooled to room temperature, and the solvents are removed under reduced pressure. To the residual off-white solids are added ether (800 mL) and deionized water (400 mL). The suspension is stirred until the solids are dissolved, and the resulting biphasic mixture is transferred to a 2-L separatory funnel. The layers are partitioned, and the aqueous phase is extracted with ether (4 x 200 mL). The organic phases are combined, dried over K2CO3 (ca. 100 g), filtered, and the solvents are removed under reduced pressure (Note 17) to afford 14.5-15.8 g (92-100%) of the pure title compound as a white solid (Note 18). (S)-1,1-Diphenylprolinol [8]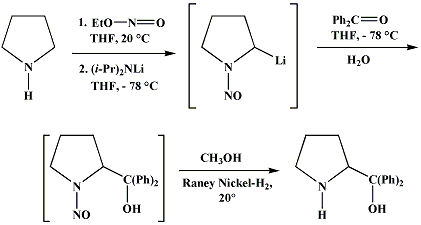 A dry, 250-ml., one-necked, round-bottomed flask equipped with a magnetic stirrer and a three-way stopcock is charged with 4 g. (0.05 mole) of ethyl nitrite (Note 1), 4 g. of dry tetrahydrofuran (Note 2), and 2.35 g. (0.0331 mole) of pyrrolidine (Note 3). The stopcock is closed (Note 4), and the mixture is stirred at room temperature for 2 days. Excess ethyl nitrite, tetrahydrofuran, and the ethanol formed are removed from the N-nitrosopyrrolidine (Note 5) by stirring at 25°C under reduced pressure (10 mm., water aspirator, (Note 6) for 2 hours. The stopcock is fitted with a rubber septum, the air in the system is replaced with dry argon (Note 4) and (Note 7)), and 50 ml. of tetrahydrofuran is injected by syringe. A solution of lithium diisopropylamide is prepared in a separate, dry, 100-ml. flask by adding 21.1 ml. (0.0340 mole) of a 1.61 M solution of n-butyllithium in hexane (Note 8) to a solution of 3.44 g. (4.76 ml., 0.0341 mole) of diisopropylamine (Note 9) in 25 ml. of tetrahydrofuran at -78°C (methanol-dry ice bath) with stirring under argon. The solution is warmed to 0°C in 15 minutes, then added dropwise with a syringe within 4 minutes to the nitrosamine solution, stirred at -78°C. Stirring of the yellow to orange solution is continued at this temperature for 25 minutes. A solution of 5.46 g. (0.0300 mole) of benzophenone in 12 ml. of tetrahydrofuran is added dropwise by syringe (Note 10), and the mixture is stirred for 12 hours at -78°C, then warmed to 0°C within 2 hours. After addition of 0.6 ml. (0.03 mole) of water, the flask is transferred from the argon line to a rotary evaporator (within the hood). Solvents and diisopropylamine are removed under reduced pressure in a 40? bath (Note 11). The remaining solid is dissolved with slight warming in 120 ml. of dry methanol (Note 12), before 3.9 g. (66 equivalents) of Raney nickel (Note 13) is rinsed into the solution with 30 ml. of dry methanol. The reaction vessel is equipped again with the three-way stopcock, and the air in the flask is replaced with hydrogen (Note 7). The flask is filled five times with hydrogen from a balloon; during this operation vigorous stirring of the Raney nickel-methanol suspension is necessary. The flask is attached to a mercury bubbler to maintain a positive hydrogen pressure (200 mm.) supplied from a cylinder, as shown in Figure 1. The reaction mixture is stirred for 3 hours at room temperature while a slow stream of hydrogen is passed through the system. The major part of the solution is decanted and filtered, and the remaining Raney-nickel suspension is extracted by refluxing three times for 10 minutes each with 20 ml. of methanol (Note 14). The combined methanol solutions are concentrated under reduced pressure. The residue is dissolved in 150 ml. of diethyl ether and 100 ml. of water, the layers are separated (Note 15), and the aqueous layer is extracted three times with 50-ml. portions of ether. The combined extracts are dried over sodium carbonate and concentrated in a rotary evaporator to a total volume of 150 ml. Dry hydrogen chloride gas is bubbled into the solution with stirring until the mixture is acidic, The almost colorless precipitate of the hydrochloride is filtered, washed two times with 30-ml. portions of dry ether, and dried in a desiccator under reduced pressure for 3 hours, giving 5.99-6.11 g. (58-60%, based on benzophenone) of the product, m.p. 244-249°C (dec.). Recrystallization from methanol?acetone gives 5.06-5.20 g. (58-60%) of analytically pure product, m.p. 267-269°C (dec.) (Note 16). The free base is obtained by treatment of the hydrochloride with 10% aqueous sodium hydroxide and extraction with ether, m.p. 82-83°C (Note 17). (S)-1,1-Diphenylprolinol [9]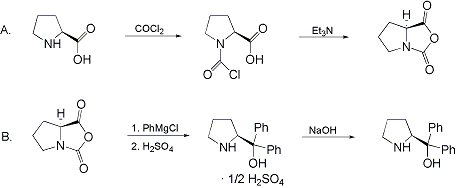 A. (S)-Proline-N-carboxyanhydride. A dry, 3-L, three-necked flask, fitted with a mechanical stirrer, nitrogen inlet tube, 500-mL, pressure-equalized addition funnel and a Teflon-coated thermocouple probe, containing dry tetrahydrofuran (THF) (600 mL) is charged with (S)-proline (60.0 g, 0.521 mol) (Note 1). To the cooled (15–20°C) suspension is added phosgene (324 mL, 0.625 mol, 1.93 M in toluene), via a pressure-equalized addition funnel, over a 0.5–1.0 hr period maintaining a temperature of 15–20°C (Note 2). The reaction mixture is then aged for 0.5–0.75 hr at 30–40°C. The reaction mixture should become homogeneous as the proline reacts with the phosgene to afford the intermediate N-carbamoyl chloride. Once homogeneous, the reaction mixture is aged an additional 0.5 hr, then concentrated under reduced pressure (15–20°C, 1000 down to 50 mBar) to a volume of 80 mL (Note 3). Dry tetrahydrofuran (600 mL, KF < 50 µg/mL) is added to the mixture and it is cooled to 0–5°C. Dry triethylamine (72.6 mL, 0.521 mol, KF < 50 µg/mL) is added over 0.25 hr (Note 4). The mixture is aged for 0.5 hr at 0–5°C, then filtered through an enclosed, 2-L, medium-frit Schlenk funnel under an atmosphere of nitrogen (with careful exclusion of moisture). The triethylammonium hydrochloride cake is washed with dry tetrahydrofuran (3 × 105 mL, KF < 50 µg/mL). The filtrate and washes are combined and used as is in the next reaction (Note 5) and (Note 6). B. (S)-1,1-Diphenylprolinol. A 3-L, three-necked flask, fitted with a mechanical stirrer, nitrogen inlet tube, 1-L addition funnel and Teflon-coated thermocouple probe, is charged with phenylmagnesium chloride (0.80 L, 1.6 mol, 2.0 M in tetrahydrofuran) (Note 7) and cooled to -10°C (Note 8). To the suspension is added, with stirring, the tetrahydrofuran solution of (S)-proline-N-carboxyanhydride over a 1-hr period maintaining the temperature at -15° to -10°C (Note 9). After the addition is complete, the reaction mixture is aged for 3 hr at -15°C, then 1 hr at 0°C (Note 10). The reaction is quenched into a 5-L, mechanically-stirred flask containing 2 M aqueous sulfuric acid (1.05 L, 2.1 mol) (0°C) over a 0.5–1.0 hr period (Note 11). The reaction mixture is aged for 1 hr at 0–5°C, filtered, and the magnesium sulfate cake washed with tetrahydrofuran (3 × 500 mL) (Note 12). The filtrate and tetrahydrofuran washes are combined and concentrated (1 atm) to a volume of 1.0 L (Note 13). The mixture is cooled to 0°C, aged 1 hr, and then filtered. The cake is washed with water (5°C, 2 × 100 mL) and ethyl acetate (3 × 180 mL) (Note 14). The cake is dried under reduced pressure (40°C, 50 mBar) to yield 79–88 g (50–56% based on proline) of (S)-diphenylprolinol sulfate as a free-flowing white crystalline solid, mp 275–290°C (Note 15). A 3-L, three-necked flask, fitted with a mechanical stirrer, nitrogen inlet tube, and Teflon-coated thermocouple, is charged with (S)-diphenylprolinol sulfate (81.0 g, 0.268 mol), tetrahydrofuran (268 mL), and 2 M aqueous sodium hydroxide (268 mL). The mixture is stirred at 20–25°C until all the solid dissolves (0.5 hr) (Note 16). Toluene (1.1 L) is added and the mixture is aged 0.5 hr. The two-phase mixture is filtered through a medium frit sintered glass funnel and partitioned (Note 17). The upper layer is washed with water (130 mL) and the toluene is removed under reduced pressure to afford a light-tan colored oil that crystallizes upon standing at ambient temperature (Note 18). Recrystallization of the resulting light-tan crystals from heptane (ca. 2 mL of heptane per gram of prolinol) affords (S)-1,1-diphenylprolinol; [yield 64–68 g (94–99% yield based upon diphenylprolinol sulfate)] as white crystals, mp 76–78°C, >98% ee (Note 19),(Note 20)). References [1] French patent 3638M
|