In 1933, Gilman showed that on carbonation phenyllithium yielded 70% benzophenone and no benzoic acid, which is the main product on carbonation of the corresponding magnesium compound. They found that the reason for the high yield of ketone was the higher reactivity of the organolithium compound. If an aryllithium compound was allowed to react with carbon dioxide at temperatures between -50°C and -80°C the following reaction occurred:
RLi + CO2  RCOOLi
RCOOLi
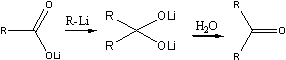
At a higher temperature (room temperature) another reaction took place. To the lithium salt of ArCOOH was added one mole of aryllithium, and a dilithium salt of a dihydroxymethane was obtained, which on hydrolysis yielded a ketone in accordance with the following general reaction:
Only in one case, hitherto, has any comparison been made between the use of the free acid or its lithium salt: In the case of benzophenone Gilman and van Ess have made two syntheses. One started with the lithium benzoate, which was allowed to react with one mole of phenyllithium, and was found to give a 70% yield of ketone and no tertiary alcohol; the second experiment started with benzoic acid and two moles of phenyllithium and yielded 37.2% of ketone and 14.1% of triphenylcarbinol.
Two possible explanations are given for the formation of the tertiary alcohol. The first is that benzoic acid is dehydrated by phenyllithium to give benzoic anhydride. This would react with one mole of phenyllithium to give lithium benzoate and "free" benzophenone, which would enter the ordinary reaction of a ketone yielding triphenylcarbinol:
2 C6H5COOH + 2 C6H5Li (C6H5CO)2O
(C6H5CO)2O + C6H5Li (C6H5)2CO + C6H5COOLi
(C6H5)2CO + C6H5Li (C6H5)3COLi -> (C6H5)3COH
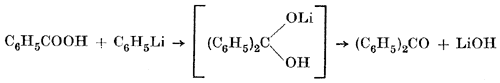
Another explanation is suggested, in which the phenyllithium is supposed to be added directly to the carbonyl bond (without previous formation of lithium salt) yielding an intermediary product (C6H5COOLi), which by phenyllithium is brought over into the alcohol in accordance with the reaction above.
When using methyllithium, however, these side reactions are not likely to occur, because of the markedly lower reactivity of methyllithium as compared with phenyllithium, unless the concentration of methyllithium is strongly increased. Karrer have shown that when one mole of β-ionylidene crotonic acid is allowed to react with two moles of methyllithium only ketone is formed. If the number of moles of methyllithium is increased to five, equal quantities of ketone and carbinol are obtained, and by a further increase of methyllithium to ten moles per mole acid, the tertiary alcohol will bo formed almost exclusively. In the case of the two higher concentrations the explanations given by Gilman seem very probable. The side reaction of carbinol formation may be avoided by adding the methyllithium solution to the acid, thus avoiding too high concentrations of methyllithium.
It appears that slightly higher yields were obtained by using the lithium salt. However, as the lithium salts in some cases are difficult to obtain, all syntheses have been carried out with the free acid.
In methyllithium there is as in other organometallics a displacement of electrons towards the carbon atom, forming a carbanion
with a free electron pair and a lithium ion: H3CLi H3C- + Li+
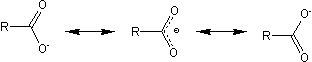
The lithium salt of a carboxylic acid has the following resonance structures:
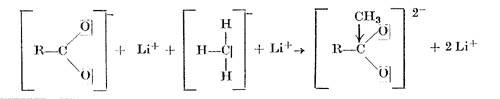
On the addition of methyllithium to the lithium salt of a carboxylic acid, the carbanion with its unshared electron pair adds to the sextet of the carboxylic acid anion, with the formation of a new carbon-carbon bond, and the oxygen with one of its unshared electron pairs forms a bond with the lithium atom The dilithium salt of the dihydroxymethane thus obtained is hydrolysed by water to give the methyl ketone.
In conclusion it may be said that the preparation of methyl ketones by the action of methyllithium on carboxylic acids generally gives excellent yields, and that no interference with double bonds occurs. As a rule acids having an electron-attracting group seem to give a higher yield of ketone than those containing a electron-repelling group.
Experimental
Preparation of methyllithium
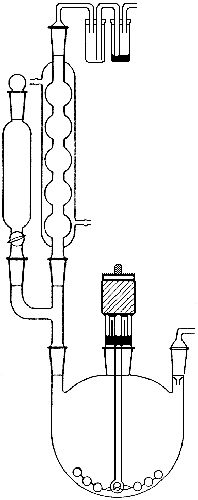
The preparation of methyllithium was carried out in the apparatus shown in Fig. 1, a three-necked flask equipped with a dropping funnel (50 ml), a reflux condenser with a mercury trap, a Hershberg stirrer with a mercury seal and a gas inlet tube.
For the preparation of methyllithium three methods were tried, one using methyl chloride and two with different ratios of methyl iodide/lithium metal. When using methyl chloride the yields varied between 20-50%.
Using the more easily handled methyl iodide Gilman give two different ratios for the concentration of methyl iodide/lithium metal, 1.25/1.0 and 1.0/2.2; both said to yield similar results. However, in the authors experiments the latter ratio has been found to give higher yields, and has therefore been used in this work. (With the ratio 1.25/1.0 the yields were 75-50%, and with the ratio 1.0/2.2 90-95% yields were obtained.)
Example of a preparation using the latter ratio
0.35 mole (2.4 g) of lithium sand, prepared according to Perrine and Rapoport, was brought into the reaction vessel with 100 ml of ether (The ether has always been dried with sodium wire). The air in the apparatus was then expelled with nitrogen gas, previously dried over calcium chloride, phosphorus pentoxide and soda asbestos ("Ascarite"). The Hershberg stirrer was started, and the stream of nitrogen was slowed down. The speed of the gas flow could be noticed at the mercury trap, which also served to give a slight overpressure to exclude moisture, oxygen and carbon dioxide.
To the lithium metal was added 1 ml of methyl iodide, and the reaction was started by carefully heating on a water bath. When the reaction began the water bath was removed and another 100 ml of ether was added, and 0.16 mole (23 g) of methyl iodide diluted with 75 ml of ether was then introduced at such a rate as to cause gentle reflux. After all the methyl iodide had been added the mixture was refluxed on a water bath for 1 hour, after which time the stirrer was stopped and the solution allowed to cool. The unreacted lithium metal then rose to the surface and a greyish white precipitate of lithium iodide and metallic lithium settled at the bottom of the flask, leaving a weakly opalescent solution between. Samples of the solution were taken and after hydrolysis with water titrated with 0.1 N hydrochloric acid. Yield 0.15 mole of methyllithium (230 ml 0.67 N methyllithium solution; 96%).
As there is no exact method of determining the concentration of methyllithium, only small amounts were prepared and were used in the coupling reaction immediately after the preparation.
Preparation of methyl ketones using the free acid
For the coupling reaction between methyllithium and carboxylic acids an apparatus was used consisting of a round-bottomed flask (250 or 100 ml) with a side tube for a gas inlet capillary and equipped with a reflux condenser protected by a sodium hydroxide drying tube. The reaction was carried out in a nitrogen atmosphere, the nitrogen gas being dried over calcium chloride, phosphorus pentoxide and soda asbestos. The coupling was carried out according to the following general scheme:
0.03 mole of the acid dissolved in 50 ml of ether was brought into the reaction vessel, and the air was then expelled with a rapid stream of nitrogen. After 2-3 minutes the gas stream was slowed down, just to create sufficient bubbles for stirring the solution during the experiment.
When all air had been expelled 0.06-0.07 mole of an ethereal solution of methyllithium was added through the condenser. A vivid reaction took place, the ether refluxed, and a white precipitate was formed (lithium salt of the acid). After the addition of all the methyllithium the precipitate sometimes dissolved and a weakly opalescent solution was obtained. If necessary, the solution was then refluxed for 10-30 minutes to complete the reaction. After the solution had reached room temperature, water was slowly added. The excess of methyllithium was thus destroyed and lithium hydroxide was formed from the intermediate dilithium salt of the dihydroxymethane.
The alkaline water layer, which contained the lithium salt of unreacted acid, was removed in a separatory funnel, and the ethereal layer washed three times with half its volume of water. The ether solution was then dried over magnesium sulphate, filtered and the ether driven off, first at ordinary pressure and then at 8-10 torr. The ketone was purified if necessary, which however was seldom the case. If needed, derivatives were prepared for characterisation of the ketone.
The acids which have been investigated were coupled with methyllithium in a series of runs. The experiments and the isolation of the methyl ketones were carried out according to the general scheme above. Any deviations from this will be given in the case of the individual acids below.
Cinnamic acid
To a solution of 0.0088 mole (1.3 g) of cinnamic acid in 50 ml of ether was added 0.018 mole of methyllithium. The yield of benzalacetone was 1.0 g. 2,4-dinitrophenylhydrazone from alcohol mp 226-227°C, yield 77%.
Benzoic acid
A solution of 0.027 mole (3.3 g) of benzoic acid in 50 ml of ether was coupled with 0.055 mole of methyllithium. On isolating the ketone, the water layer was extracted twice with 50 ml of ether and the ethereal solutions were treated according to the general scheme. 2.7 g of acetophenone was isolated; yield 82%.
Phenylacetic acid
0.026 mole (3,5 g) of phenylacetic acid was allowed to react with 0.055 mole of methyllithium. A white precipitate separated, which partly dissolved. Isolation gave 2.65 g of phenylacetone; yield 76 %.
Preparation of methyl ketone using the lithium salt
Cinnamic acid was dissolved in the calculated amount of 1.98 N lithium hydroxide in water. The solution was evaporated to dryness and the solid residue finely divided in a mortar. The lithium salt was then washed with ether and dried at 120°C for 10 hours. To a suspension of 0.013 mole of finely divided lithium cinnamate in 25 ml of ether was added 0.015 mole of an ethereal methyllithium solution. On refluxing for 2 hours the main part of the salt had reacted. Isolation (as above) of the ketone gave 1.3 g of benzalacetone; yield 69 %. In another run the time of reflux was increased to 6 hours. In this case the yield was 79%.