Enolate phenylacetone synthesis FAQ 1.0by Drone #342So as to ensure that nobody thinks I’ve been shirking in my work here, I thought I’d post some gleanings from the enolate literature I scanned in a couple weeks ago. I dedicate this FAQ to the lovely and delightfully lecivious Fraeulein, ms honey. Phenylacetone is a nifty chemical; much sought after, but oh-so-elusive due to silly little societal restrictions. But enough political diatribing; let’s get to the matters at hand -- how to make it out of acetone, and common chemicals. There are a myriad of ingenious methods of making phenylacetone – some starting with phenylacetic acid, some with benzyl chloride, and dozens of others. This FAQ will concern itself with producing this lovely stuff from halobenzenes (i.e. chlorobenzene, bromobenzene, and iodobenzene), and acetone enolates – made by reacting acetone with a strong base. I won’t go into the mechanisms and enolate chemistry too far, other than a general overview. Enolate chemistry is a fascinating field – much of it very cutting edge (though this ain’t.) Generally, what we have here is a strong base (i.e. a base with a pKa higher than acetone, which is 23), reacting with acetone to form an enolate: 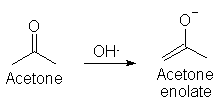
This is a great nucleophile, and the actual reaction that makes the good stuff is a simple nucleophilic Sn1 Reaction, where the halide dissociates from the halobenzene, leaving it open for an attack by the enolate, which produces phenylacetone and a salt. But here’s the other thing to take into consideration. The way that the halobenzene reacts is that the halogen has to come off first. This means that the better the halogen is at leaving, the faster everything goes. In DMSO, iodobenzene is about 6 times faster than bromobenzene(1), and no real study has been for chlorobenzene that I’ve found, though there have been studies using it in this type of reaction. We’ll just leave fluorobenzene alone. Anyway, any college chemistry book will describe these things in much better detail and with a lot more clarity than I can right now (I’m in a hurry.) Now, let’s get to business. Phenylacetone from acetone enolate is a reaction that’s been very well covered, since its such a classic, simple Sn1 reaction to study. The reagents are cheap, and its all pretty straight forward. Because of this, a ton of variations have been investigated, and a lot have progress has been made. The kinetics are all well-studied, catalysts have been investigated, solvents, different halides, etc. This leaves us with a lot of good stuff to work with. Rather than re-invent the wheel, I decided I’ll do a review of the best articles, and include the relevant experimental sections. Enjoy; may you never sleep. JOC, 42, 8, 1977, 1449-1457 This is the one that started it all, essentially. The title was "Dark Reactions of Halobenzenes with Pinacolone Enolate Ion. Evidence for a thermally induced Aromatic Snr1 Reaction". In this reaction, they used various catalysts in different quantities to get this going a little faster. Typical procedures included 0.024 M PhI, 0.1 M enolate, and a catalyst like bubbling O2 or 0.0025 M Fe(NO3)3. Reactions were essentially done after an hour. There are a few things of course to keep in mind here. Of course, acetone should be substituted in equimolar amounts wherever they use pinacolone. Everything should be anhydrous, and that’s mainly it. "Dark Reaction of Halobenzene and Pinacolone Enolate in Me2SO. Typical Procedure. A. Potassium tert-butoxide (freshly sublimed) was placed into a 100-ml one-neck flask with a gas inlet side arm and short condenser. The flask and fittings were completely wrapped with opaque tape and all reactions were carried out in a nitrogen atmosphere. Dimethyl sulfoxide (50 ml) was added with a syringe through the condenser and after several minutes of stirring the apparatus was placed in a 25 *C thermostat bath. After equilibration the appropriate amounts (Table 1) of aryl halide and pinacolone were added and the flask was well swirled to mix the contents. At the desired time (Table 1), the solution was acidified with dilute nitric acid, 150 ml of water was added, and the mixture was extracted with three portions of ether. The combined ether fractions were washed with water and dried. In preparative runs, the ether was removed and the resulting product was purified by preparative GLC and distillation. In analysis runs, an internal standard was added and the ether solution was analyzed by GLC. The aqueous extracts were usually titrated for halide ion." JOC 1983, 48, 3109-3112 The procedure they refer to by Bunnett, Scamehorn, and Traber is the procedure in JOC, 42, 8, 1977, 1449-1457 (see earlier in this FAQ.) Using the modifications they list for bromobenzene gives by far the most attractive conditions. "Reaction of Acetone Enolate (1) and Iodobenzene (2). The procedure followed was similar to that described by Bunnett Scamehorn, and Traber. 7 The deep-brown solution obtained by mixing acetone enolate (10 mmol) with iodobenzene (2.4 mmol) was irradiated for 1 h (3.8 h when filtered) with a focused high pressure mercury lamp. The reaction was quenched with 60 ml of water and extracted three times with ether. The organic layer was analyzed by GLC (5% SE30 on Chromosorb P. 130-230 °C at 4°/min, biphenyl internal standard). The major product isolated was phenylacetone (88%), although a number of unidentified minor products could be detected in trace quantity. "Reaction of Acetone Enolate (1) and Bromobenzene (2). In a procedure similar to that described above, the enolate derived from 0.73 ml (10.0 mmol) of acetone was allowed to react with 0.25 ml (2.4 mmol) of bromobenzene. After irradiation of the bright yellow charge transfer complex with a high-pressure mercury lamp for 1.25 h (6 h filtered), the solution became orange. After workup as described above, phenylacetone was obtained in 94% yield." JOC, 1984, 49, 4881-4883 …Just another example of the same reaction involving iodobenzene and acetone enolate in DMSO. Substitute acetone for pinacolone accordingly. Yields of the acetone are around 50% from iodobenzene. "Reaction of Iodobenzene and Ketone Enolate in Me2SO. General Procedure. Me2SO (25 ml) was transferred by syringe into a N2-purged 100-mL 3-neck flask fitted with two stoppers and a closed-end 12-mm tube with a 60° bend. The flask and tube were wrapped with black opaque tape. Freshly sublimed potassium tert-butoxide (1.22 g, 0.010 mol) was added, and the tube was charged with 1.00 g (0.010 mol) of pinacolone and 0.51 g (0.0025 mol) of iodobenzene. Stirring was employed to dissolve the base, and the tube containing the ketone and PhI was cooled with a dry ice acetone bath The system was evacuated and filled with nitrogen. This procedure was repeated 3-8 times. After the freeze pump-thaw cycles were complete, the reactants were added to the flask by rotation of the bent tube. The solution was stirred and the flask placed in a 25 °C temperature bath. After 1 h, 6 N sulfuric acid (1.85 ml) was added. The solution was diluted with 50 ml of water and extracted 3x with ether. The combined ether extract was washed with water (3x) and dried (MgSO4), and an internal standard (phenylacetone) added for GLC analysis. The aqueous layers were combined and an aliquot was used for iodide analysis. For each ketone studied, the product from at least one reaction was isolated by preparative GLC or by column chromatography on silica gel, and the IR and NMR spectra were compared with those of authentic compounds. Four identical experiments carried out as above with iodobenzene and pinacolone gave yields (r) of 67.2%, 60.0%, 66.5%, and 68.1%." JOC, 1984, 49, 3041-3042 Now this one is interesting for several reasons. First, the main point of research was finding catalysts to activate the halobenzene for nucleophilic attack. As it turns out, FeSO4 worked pretty well for reactions involving iodobenzene, and even involving chloroaniline (yield was 51%, though it undergoes an intramolecular reaction to form 2-methylindole, which was the chemical measured to determine yield), though mixed results were obtained with bromobenzene. Its important to note that results were best when ANHYDROUS FeSO4 was used. The other interesting things was that they used ammonia as their solvent. Why? I guess they wanted to go with a classic, though DMSO is a lot nicer to deal with. Maybe they didn’t want DMSO already seemingly catalytic effect interfering. Who knows? Anyway, here it is. "Typical experimental procedure: Ammonia (60 ml) was distilled from sodium into a three-necked flask flushed with N2 and equipped with a cold-finger type condenser (with 2propanol and solid CO2 in the coolant well) and a serum cap. The glassware had been flamed in a stream of nitrogen. Under magnetic stirring,4.5 g of freshly sublimed t-BuOK and 300 mg of dried FeSO4 were added through paper funnels. The color of the solution became green-gray. Pinacolone (3.8 g) was added from a syringe and after few minutes also PhI (2.6 g) was added from a syringe This marked the beginning of the reaction. The flask was kept under magnetic stirring in a dark hood for 20 min. and then NH4NOS was added to quench the reaction. A rusty color developed; after evaporation of ammonia, water was added and the mixture was extracted with ether, washed with water and dried over Na2SO4. Removal of the solvent left a yellow liquid which was distilled (bp 98 °C at 2 mm Hg) to give 1.9 5 of colorless liquid 2 (85% yield)." Well, that’s about it for now. Look for future FAQ’s, with more user-friendly directions than this one, as well as directions for producing your own home-made bromobenzene and iodobenzene from OTC materials. This ought to give you enough to gnaw on for a long while. I've written a lot about practical clandestine enolate chemistry, but there's always been a bit of a sticking point in much of my discussion. How does one practically make enolates at home? Enolate chemistry often requires *strong* bases, ones that mere WATER is far to acidic to have in its presence (pKa>15), so what's a bee to do? Alkoxides.By mixing sodium metal with an anhydrous alcohol, the sodium with strip off the hydroxy's relatively acidic proton, making an alkoxide -- a base of great strength and usefullness in making enolates at home. Just to give you an idea regarding their acidity, NaOH has a pKa of about 14, NaOMe has a pKa of 15, NaOEt's is around 26, and NaOiPr around 28-30 (all of this is measured in DMSO, and the numbers kinda vary from source to source.) Here's how. Now its important that everything be absolutely anhydrous, and some saferty precautions should be taken. The alcohol must be 99+% pure, the glassware should be flame-dried. Good ventilation is needed, since the reaction generates hydrogen gas, and having a build-up of that can lead to Hindenburgian results. With that said, let's get cookin'! In a 250-mL RB flask equipt with a reflux column and a magnetic stirrer, nestled in an ice bath, 25 mL of acohol (MeOH, EtOH, iPrOH) is allowed to chill with stirring. Once the alcohol is good and chilled, 1.32 grams of cleaned sodium metal is carefully added, and a tube full of dried CaCl2 to placed on the outlet of the relux column. Immediately, the reaction begins to take place, and a lot of heat and hydrogen are generated. THe solution will begin to boil as the sodium dissolves, and eventually, when all the melodrama of the disolving is over, replace the icebath with a hot water bath, and reflux an additional 15 minutes. From there, evaporate off the excess alcohol, yielding the final product (around 3.5 grams.) Scaling up requires a little extra finess. When using EtOH or iPrOH, chilling the alcohol first in an acetone/dry ice bath, then gradually allowing the solution to come to room temperature will allow you to add the sodium metal without too many fireworks. Always keep ventilation a top priority. The final product will be white-yellow (with ethoxide, its very yellow), and decomposes with too much exposure to air. So, once you have a nice, off-white mass of crap, keep it tightly sealed right up until you need to measure it. |