Regioselective Oxyiodination of Aromatics Using KI/OxoneSynth. Comm. 32(15), 2319-24 (2002)[ Back to the Chemistry Archive ]Aryl iodides have widely been successfully used as synthetic intermediates in the formation of new carbon–carbon bonds via replacement of their iodine atoms with electrophiles. [1a-h] For more than a century, e.g., in the synthesis of Fanta [2] and Heck [3], more extensive progress has retarded due to the lack of simple and reliable methods of their preparation [4]. However, synthetic methods leading to aromatic iodides [5] by direct iodination of aromatic compounds are relatively limited compared with those of preparing aromatic chlorides and bromides because of the low electrophilicity of iodine. Although halogenation of aromatic compounds with halogens is well known reaction, bromination and chlorination easily proceed with or sometimes without Lewis acid catalysts, but iodination usually more difficult to take place. Except for active substrates, oxidizing reagents such as nitric acid, iodic acid, sulfurtrioxide, sulfuric acid and hydrogen peroxide must normally used in order to generate a better electrophile6 by oxidation of molecular iodine. It was also reported that the aromatic iodination with molecular iodine was catalyzed by stoichiometric amounts of metal halides, such as AlCl3–CuCl2 [7], or SbCl5 [8], and a direct iodination was carried out by using NH4l catalytic amounts of NOBF4 in CF3COOH/CH2Cl2 or CF3COOH/CH3COOH with molecular oxygen [9]. These procedures suffer some deficiencies. The most serious of these are substrate limitations due to reaction conditions and the loss of iodine from the reaction as hydrogen iodide or metallic iodide. We have designed a novel system to generate electrophilic iodine in situ from easily available KI as an iodine source, commercially available Oxone® as an oxidant for the oxyiodination as a possible alternative to solve the disadvantages described in the earlier methods. In this communication we report a new method for the para selective oxyiodination of aromatic compounds using Oxone® as an oxidant, and KI as an iodine source. 
Potassium peroxymonosulfate is an inexpensive and readily accessible oxidizing agent. It is commonly used as Oxone® (2KHSO5.KHSO4.K2SO4) and is a versatile oxidant for the transformation of a wide range of functional groups [10]. A number of different aromatic compounds were subjected to the iodination reaction to test the generality of this method and the results are presented in Table 1. These reactions proceeded efficiently under mild conditions in methanol with high yields and regioselectivity with KI and Oxone®. As Table 1 shows that the reaction gives high yields and para-selectivity for a range of substituted benzenes of high activity. The results in Table 1 indicate that activated aromatic compounds are more selective for nuclear iodination. Introduction of an electron-withdrawing group on the aromatic ring substantially decreases the rate of ring iodination (Table 1, Entries 2 and 4) while on electron donating group increases it. 2-methoxy-naphthalene gives 1-iodo-2-methoxy naphthalene (Scheme 1). When the less reactive aromatics such as nitro benzene, benzoic acid failed to undergo iodination under the same reaction conditions. (1) ArH + KI + 2 KHSO5·KHSO4·K2SO4 -> ArI + KOH + K2S2O8·KHSO4·K2SO4 + H2O (2) 2 KHSO5·KHSO4·K2SO4 + KI -> KOH + HOI + K2S2O8·KHSO4·K2SO4 (3) 2 HOOSO3K -> 2 ·OH + 2 ·OSO3K (4) KI + 2 ·OH + ·OSO3K -> KOH + HOI + K2S2O8 (5) ArH + HOI -> ArI + H2O A wide range of solvents has been used in these reactions including carbon tetrachloride, hexane, dichloromethane, methanol and acetonitrile. The best results were obtained by using methanol as a solvent compared to others. We surveyed the iodination with various oxidants such as Oxone®, tert-butylhydroperoxide, hydrogen peroxide and molecular O2. However, Oxone® is far superior to the other oxidants. The role of Oxone® was confirmed by conducting a blank experiment where the formation of iodo compounds was not observed. A typical oxyiodination of an aromatic compounds in the presence of Oxone® proceeds according to the stoichiometry of Eq. (1). It is believed that the iodination proceeds via the formation of hypoiodous acid. The hypoiodous acid has higher instability due to pronounced ionic nature and thus more reactivity towards the aromatic nucleus. The absence of iodination of the ring methyl group (Table 1, Entries 11, 12 and 13) is indicative of the electrophilic mechanism of the reaction rather than a radical pathway. Oxone®, in direct comparison, has a higher onset of decomposition than hydrogen peroxide and liberates less energy. This reaction is performed at lower temperature, which provides a larger margin of safety. Additionally Oxone® is a solid, allowing for the addition of precisely weighed amounts of reagent to be used in the reaction. In conclusion, we have developed a novel system using Oxone® as an interesting alternative to hydrogen peroxide in the oxidative iodination of aromatic substrates. The commercial availability of the reagents, simple reaction conditions, no evolution of hydrogen iodide, high yields of monoiodinated products and work-up is simple. The absence of side chain iodination products in reaction conducted in methanol suggests a substantial increase in the rate of the ionic process. General Procedure for the iodination of aromatic compoundsOxone® (2.2 mmol) was added to a well stirred solution of KI (2.2 mmol) and substrate (2 mmol) in methanol (10 mL) and the reaction mixture was allowed to stir at room temperature. The reaction was monitored by thin layer chromatography (TLC). After the completion of the reaction, the mixture was filtered and solvent evaporated under reduced pressure. The products were purified by column chromatography over silica gel and confirmed by 1H NMR and mass spectra. 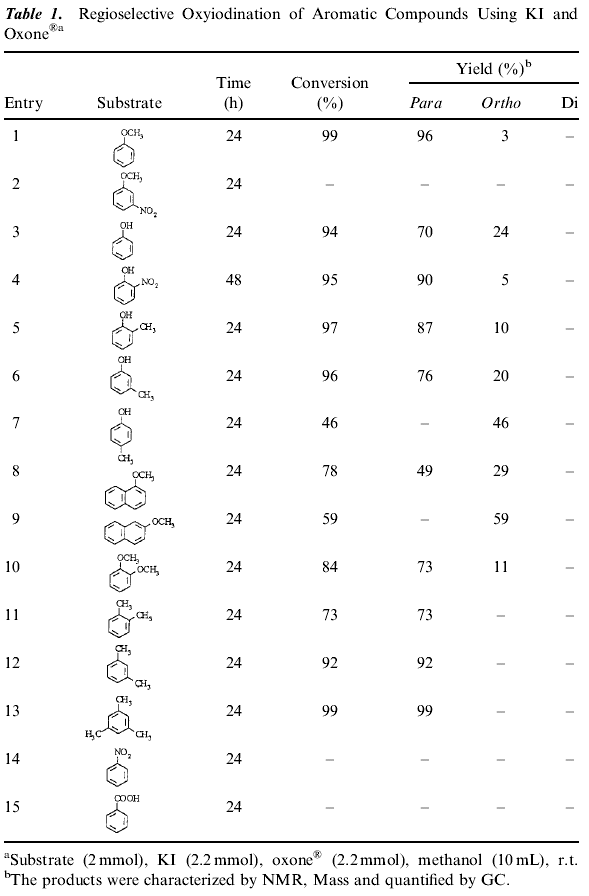
References [1] 1a Fanta P.E., Synlett., (1974) 9., 1b Nilsson M., Tetrahedron Lett., (1966) 679., 1c House H.O., Respess W.L., Whitesides G.M., J. Org. Chem., 31 (1966) 3128., 1d Matsui K., Tobita E., Ando M., Kondo K., Chem. Lett., (1981) 1719., 1e Burdon J., Coc P.L., Marsh C.R., J. Chem. Soc. Chem. Commun., (1967) 1259., 1f Setsune J., Matsukawa K., Wakemoto H., Kitano T., Chem. Lett., (1981) 367., 1g Suzuki H., Kobayashi T., Yoshida Y., Osuka A., Chem. Lett., (1983) 193., 1h Suzuki H., Abe H., Osuka A., Chem. Lett., (1980) 1363.
|