Abstract
Nitration of cyclic conjugated olefins was achieved in a one-pot procedure using nitryl iodide generated in situ from iodine and potassium nitrite complexed with 18-crown-6 in tetrahydrofuran under sonication/stirring, followed by treatment of the unstable iodo nitro compound with base. The yield of the nitro compound varied from 52 to 90%.
Conjugated nitroolefins have been recognized as versatile synthetic intermediates in various organic syntheses because of their easy conversion to a variety of diverse functionalities1. Several methods are available for the preparation of nitroolefins1-5. However, much demand still exists for a method to prepare nitroolefins in a convenient and efficient way.
In our ongoing effort to synthesize novel 2-aminotetralins as potential biologically active agents6, we needed an efficient method for the preparation of the key intermediates, cyclic conjugated olefinic nitro compounds. However, attempts to prepare those nitroolefins in acceptable yield using available methods were unsatisfactory. In this report, we present a general, mild, and practical method for the nitration of olefins.
This method is based on the regioselective addition of nitryl iodide to olefins followed by base induced bimolecular elimination of iodide to form a nitroolefin. Previously, Hassner and coworkers (1969) elucidated the mechanism of formation of nitroolefins via an unstable iodo nitro intermediate7. The formation of the iodo nitro adduct was found to be the key step in the reaction sequence. Nitryl iodide is generally prepared by the reaction of AgNO2 and iodine as described by Birckenbach in 19328. Replacing AgNO2 with inexpensive NaNO2, Jew et al.5 reported an improved yield of nitration with NaNO2/H2O/I2/EtOAc/ethylene glycol. However, the major problem in this reaction was the presence of water as a solvent for NaNO2 in the biphasic system. This led to formation of the hydroxy nitro compound as a major side product. In a similar nitryl iodide mediated nitration, Sy and By4 observed that poor yield of nitroolefin was attributable to degradation of the unstable iodo nitro intermediate which was sensitive to moisture and oxygen. In our experiments using the NaNO2 method, we also noted the significant formation of hydroxy nitro compound, 2d (X=OH), when 1d was used as the substrate.
We envisaged that the poor solubility of nitriles in anhydrous organic solvents was one of the problems in achieving an acceptable yield. To circumvent this problem, a more organophilic nitrite would be necessary. Since potassium ions can be complexed by 18-crown-69, a phase-transfer agent, we used the KNO2/18-crown-6/I2/THF system as an alternative to the other published methods to generate nitryl iodide (Scheme). The solubility of the nitrite ion was further increased by exposing the reaction mixture to ultrasound10. Generally, the formation of the intermediate iodo nitro compound takes from a few hours2,4 to several days5. We found that the rate of reaction was significantly increased with the use of ultrasound (Table, entries 3, 4, 6, 7).
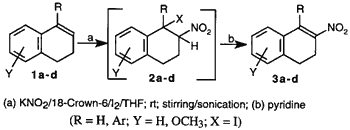
This technique appears to be an excellent, mild, regioselective, non-aqueous, one-pot procedure for the preparation of conjugated nitroolefins. When compared with earlier methods2,5, the yields from these reactions were consistently higher, particularly with sterically hindered substrates such as 1b, 1c, 1d, and 1-phenylcyclohexene. The results are summarized in the Table.
Table.
Nitration of Olefins in the Presence of a Phase Transfer Agent
| Entry | Compound/Product | R | Y | Conditionsa | Yieldb (lit.) |
| 1 | 1a/3a | H | H | PEG-400; 2.5 h; ))) | 73% (78%5) |
| 2 | 1a/3a | H | H | 18-C-6, 1.5 h; Stirring | 90% (78%5) |
| 3 | 1b/3b | Ph | H | 18-C-6, 29 h; Stirring | 68% |
| 4 | 1b/3b | Ph | H | 18-C-6, 2h; ))) | 68% |
| 5 | 1c/3c | Ph | 6,7-(MeO)2 | 18-C-6, 3 h; ))) | 52% |
| 6 | 1d/3d | 3,4-(MeO)2Ph | H | 18-C-6, 49 h; Stirring | 68% |
| 7 | 1d/3d | 3,4-(MeO)2Ph | H | 18-C-6, 4h; ))) | 68% |
| 8 | Indene/2-Nitroindene | 18-C-6; 3h; Stirring | 0%c (54%d,2) | ||
| 9 | 1-Phenylcyclohexene/ 1-NO2-2-Ph-cyclohexene | 18-C-6, 1 h; ))) | 79% (49%5) | ||
| 10 | Styrene/ trans-β-Nitrostyrene |
18-C-6; 0.5 h; Stirring | 84% (82%2, 81%5, 45%4) |
Notes
a. All the reactions were performed at r. t., the time represents the reaction time of
olefin and nitryl iodide; in all cases nitryl iodide was generated in situ with sonication.
b. Isolated yield of pure product.
c. A polymerized mixture resulted from base catalyzed self-Michael addition of nitroindene11.
d. The reaction was performed in a non-basic medium.
This method could also be used for large scale preparations. Preliminary experiments with the quite inexpensive potassium ion complexing agent, PEG 4009, showed that replacement of rather costly 18-crown-6 with this cheaper, non-toxic, and readily available polyethylene glycol in the reaction system had little negative effect on the yield or the rate of the reaction. However, further work is necessary to explore fully the use of PEG 400 in large scale preparations.
In summary, the present method provides a practical procedure for the synthesis of tri- and tetra-substituted cyclic conjugated nitroolefins in a non-aqueous mixture using phase transfer agents and ultrasound to increase the rate and yield of the reaction. We used various amounts of iodinating agent (1.24 to 3.29 equiv) to optimize the yield.
Experimental
All starting materials and reagents were purchased from Aldrich. Compound 1c12 was prepared by the reaction of PhLi/CeCl3 complex13 and 6,7-dimethoxy-1-tetralone followed by dehydration of the resulting tertiary alcohol in toluene at reflux in the presence of Amberlyst-15 cation exchange resin14. This method was an improvement (81%) over the best available method (45%)15. Compound 1d15 was prepared by Li/Br exchange between 4-bromoveratrole and BuLi at -78°C followed by reaction with freshly distilled 1-tetralone in hexanes and dehydration of the tertiary alcohol using Amberlyst-15. Compounds 1c and 1d gave satisfactory spectral data.
Nitroolefins 3; General Procedure:
A mixture of 18-crown-6 (528.6 mg, 2.0 mmol), KNO2 (2.6 mmol), and anhyd THF (5 mL) was sonicated16 at r.t. under N2 for 5 min. To the above mixture, iodine crystals (2.75 mmol) were added and sonication was continued for 30 min. The dark reaction mixture was then treated with a solution of olefin (1 mmol) in anhydrous THF (1 mL) and pyridine (0.3-0.5 mL). After sonicating or stirring for the specified time (or until the disappearance of the starting material) under N2, Et3N (1 mL) was added and stirring was continued for another 30-120 min. The solvent was evaporated under reduced pressure. The residue was dissolved in CH2Cl2 (25 mL) and washed with dil. aq. Na2S2O3 solution (2x50 mL). The organic phase was dried (MgSO4), filtered (Celite), and evaporated to yield a dark residue. Flash column/centrifugal plate chromatography on silica gel with 5-20% EtOAc in hexane as eluent yielded the nitroolefin.
2-Nitro-3,4-dihydronaphthalene (3a)
Following the above procedure, 1,2-dihydronaphthalene (119 mg, 0.91 mmol), KNO2 (220 mg, 2.58 mmol), 18-crown-6 (661 mg, 2.5 mmol), and iodine (688 mg, 2.71 mmol) gave a brown oil. Flash chromatography on silica gel with 20% EtOAc/hexane gave 143 mg (90%) of pure product; mp 49-50°C (Lit.17 mp 52°C).
3b: mp 88-90°C (Lit.18 mp 108°C).
3c: mp 148-150°C.
2d: mp 152-154°C.
3d: mp 152-154°C.