Nitration of Propenylbenzenes with Nitryl Iodide[ Back to the Chemistry Archive ] IntroductionNitryl iodide (INO2) is a particularly effective general reagent for the synthesis of phenyl-2-nitropropenes from propenylbenzenes. Other methods of doing the same transformation include direct nitration of the alkene with the explosive compound tetranitromethane, or by reacting the alkene with N2O3 to form a pseudonitrosite, which can be hydrolyzed by a base to afford the desired phenyl-2-nitropropene. The nitryl iodide used in this reaction is generated in situ by the reaction of sodium nitrite [1] or silver nitrite [2] with iodine. The mechanism is a regioselective addition of nitryl iodide across the double bond of the propenylbenzene to form the phenyl-1-iodo-2-nitropropane, which undergoes elimination of HI to give the desired phenyl-2-nitropropenes. The reaction is general for all propenylbenzenes and styrenes, though the yields are variable between about 45-80% using both methods depending on the substrate. The inert atmosphere (Nitrogen or Argon) is absolutely necessary. Yields are about half without it. Simply take a balloon of N2 and stick it on the mouth of the rxn flask and secure with wire ties. Method 1 (Using NaNO2) [1]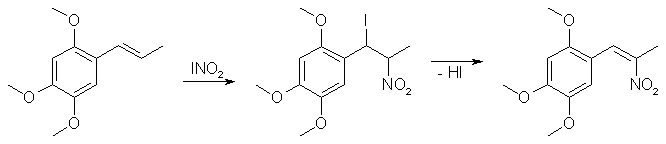
To a solution of sodium nitrite (13.24 g, 192 mmol) and ethylene glycol (8.93g, 144 mmol) in 20 mL of water, a solution of asarone (10g, 48 mmol) in 150 mL of ethyl acetate was added followed by addition of iodine (18.28g, 72 mmol) at 0°C. The reaction mixture was stirred at room temp for 48 hrs under nitrogen and then was poured into a seperatory funnel together with ethyl acetate and partitioned. The ethyl acetate solution was succesively washed with water, aqueous 10% thiosulfate and aqueous saturated NaCl. After drying over anhydrous magnesium sulfate, the ethyl acetate solution was evaporated to to obtain the crude product as dense dark yellow crystals, containing some unreacted asarone. The product was recrystallized from alcohol to afford 2,4,5-trimethoxyphenyl-2-nitropropene (2-nitroasarone) in 75%+ yield. Method 2 (Using AgNO2) [2]Iodine (1016 mg, 4 mMol) and AgNO2 (616 mg, 4 mMol) were stirred in anhydrous ether (20 ml) at room temperature under nitrogen for 45 min. Propenylbenzene (236 mg, 2 mMol) and pyridine (632 mg, 8 mMol) in ether were added and the mixture was stirred at room temp for 31 h; after this time the yellow solid was removed by filtration. The filtrate was treated with 0.5 ml Et3N and evaporated to dryness. The residue was then treated with Et3N (1 ml) in CH2Cl2 (1 ml) at room temp for 1 h. The resulting solution was evaporated to dryness, the residue was dissolved in CH2Cl2, washed with 5% aqueous NaHSO3, 5% aqueous HCl, saturated aqueous NaHCO3 and water, dried over Na2SO4 and solvent removed under reduced pressure to give a dark brown liquid. This material was chromatographed on silica and eluted with 8% ether in hexane to give the pure product (249 mg, 76.4%). The solubility of the beta-nitrostyrene product in the reaction medium is a major factor affecting the yield realized: Beta-nitrostyrenes which are relatively insoluble in the reaction medium co-precipitate with the reagent, forming a solid cake which inhibits further reaction. In such a circumstance, changing to a more suitable solvent such as THF is necessary to ensure a respectable yield. Table 1:
Beta-nitrostyrene from styrene/AgNO2/I2 [3] Silver nitrite (2.96g, 19.2 mmol), Iodine (9.75g, 38.4 mmol) were stirred in 75ml ether for 30 min, and styrene (2.00g, 19.2 mmol) was added at once, while the headspace in the reaction flask was flushed with nitrogen (necessary, or the yield will be halved), and the mixture was stirred for 4h, the ether was evaporated, and 10ml pyridine in 10ml ether was added and the mixture stirred for 2h at room temp, and then extracted with a large volume of pentane and water. The pentane was repeatedly washed with water, then dried over MgSO4, and distilled at 0.5 mmHg. The material boiling at 80-120°C was collected and recrystallized to give 1.39g (49%) of beta-nitrostyrene, mp 54°C. One-pot selective synthesis of ß-nitrostyrenes from styrenes, promoted by Cu(II)[4] We investigated the nitration of a number of substituted styrenes, and found that the procedure tolerates such functional groups on the aromatic ring as alkyl, aryl, alkoxy, acyloxy and chlorine, and can also be applied to derivatives substituted on the vinyl group. In all cases, acceptable to good yields (31-72%) of the corresponding nitrostyrenes (with a trans relationship between the phenyl and nitro groups) were obtained. alpha-methylstyrene gave a mixture of E- and Z-isomers in a ratio of 6:1. In all the cases tested, some polymeric material was obtained and the products needed to be purified by column chromatography on silica gel. No ring nitration products were detected. In summary, we have developed a convenient, inexpensive and efficient 'one-pot' operation for the selective nitration of styrenes to the corresponding ß-nitrostyrenes under mild conditions. Typical procedure for conversion of styrenes into ß-nitrostyrenes: Acetonitrile (20 ml) and NaNO2 (1.66 g, 24 mmol) were added to a solution of copper(II)tetrafluoroborate, prepared from 35% aq. HBF4 (1.62 ml, 8 mmol) and CuO (0.32 g, 4 mmol). After the mixture had been stirred for 2 min, iodine (1.52 g, 6 mmol) and the styrene (20 mmol) were introduced into the reaction flask and the mixture was stirred at room temperature for 7 h. The precipitated copper(I)iodide was filtered off after addition of water (25 ml) and the filtrate was extracted with CH2Cl2 (3 x25 ml), washed with 5% aq. sodium thiosulfate (25 ml), dried over anhydrous Na2SO4, and evaporated under reduced pressure. The resulting crude materials were purified by column chromatography (silica gel; hexane: diethyl ether, 9:1). Nitration of Isosafrole with Tetranitromethane [5] To a solution of 8.1g isosafrole and 4.8g pyridine (1.2 eqv) in 20ml acetone cooled in ice, a solution of 9.8g tetranitromethane (1 eqv) in 10ml acetone was added dropwise with good stirring. The first drop causes darkening of the reaction mixture, and after the addition is complete, the solution is opaque and red-brown. The tetranitromethane smell is soon gone, and after 2h standing in the ice-bath the dark red solution is diluted with 100ml water with good stirring, ether is added to form a top layer, and 6.7ml 33% KOH diluted to 50ml with water is added in small portions, without interrupting the stirring. The stirring is continued until all the formed pyridine-nitroform (a dark red oil) is gone. The organic layer is separated, the aqueous layer extracted with ether, and the pooled organic layers are washed with water, 5% sulfuric acid and once again with water. After removal of the ether under diminished pressure, 2-nitroisosafrole (3,4-methylenedioxyphenyl-2-nitropropane) crystallizes in yellow needles, which after recrystallization from 65ml boiling ethanol is isolated in a yield of 7g, mp 98°C. Through concentration of the mother liquor, another 0.2g is obtained. Total yield 72.5% of theory. References [1] Chemistry Letters 1747 (1986)
|