Table.
Hydroxylamines from α,β-Unsaturated Nitroalkenes
| Nitroalkene | Hydroxylaminea | Yieldb |
mpc |
| 1-Nitrocyclohexene | N-Cyclohexyl- | 78% (85) | 135-137°C |
| β-Nitrostyrene | N-(2-Phenylethyl)- | 79% (80) | 84-85°C |
| 1-Phenyl-2-nitropropene | N-(Phenylisopropyl)- | 72% (74) | 59-60°C |
| 1-(p-Br-Phenyl)- 2-nitropropene |
N-(p-Br-Phenyl- isopropyl)- | 68% (78) | 61-62°C |
| 1-(2,3-MeO-Phenyl)- 2-nitropropene |
N-(2,3-MeO-Phenyl- isopropyl)- | 63% (70) | 65-66°C |
Notes:
In view of the fact that BH3·THF is not readily available in all laboratories, we sought to develop a simpler procedure for the reduction of nitroalkenes to hydroxylamines.1 Initial experiments employing one molar equivalent of in situ generated BH3 in THF were slow and complete consumption of nitroalkene required 24 hrs. However, use of excess hydride resulted in a rapid reaction (1 hr). Fortunately the hydroxylamine precursor was stable under these conditions and no reduction to the corresponding amine2 was observed. The reduction is carried out by simply adding α,β-unsaturated nitroalkene to a stirred suspension of boron trifluoride etherate and sodium borohydride in THF at room temperature. In contrast to the previously reported borane reduction,1 no sudden exothermic reaction is observed and good yields of pure hydroxylamines are obtained (Table).
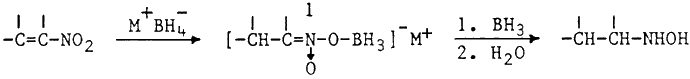
Experimental
All glassware was thoroughly dried in an oven and cooled under dry nitrogen before use. THF was dried over CaH2, distilled from LiAlH4 and stored under dry nitrogen. Commercial reagents, BF3-Et2O, 1-nitro-1-cyclohexene and ß-nitrostyrene (Aldrich) were used as received. Other nitro compounds were prepared via published procedure.3
Synthesis of N-(Hydroxyphenyl)ethylamine. General Procedure.
A flame-dried, nitrogen-flushed, 100 ml flask, equipped with a septum inlet, magnetic stirring bar and reflux condenser was cooled to 0°C. Sodium borohydride (6.3 mmol, 0.24 g) was placed in the flask followed by sequential addition of THF (10 ml) and BF3-Et2O (8 mmol, 1 ml) at 0°C. After the addition, the ice bath was removed and the contents were stirred at room temperature for 20 min. The solution of ß-nitrostyrene in THF (2 mmol, 0.3 g in 5 ml THF) was then injected dropwise into the reaction flask via a syringe. The reaction was allowed to proceed at room temperature for 1 hr and quenched by the careful addition of ice (5 g). Most of the THF was removed on a rotary evaporator, the reaction mixture acidified (1N HCl, 20 ml) and then heated at 80-90°C (oil bath) for 2 hrs. After cooling to room temperature, the acidic layer was washed with ether (2x20 ml) and then the hydroxylamine liberated via the addition of aqueous sodium hydroxide. Solid sodium chloride was added and the product extracted into ether (3x25 ml). The combined ethereal extracts were dried over anhydrous MgSO4 and the solvent removed under reduced pressure to yield 0.22 g (79%) of N-hydroxylphenylethylamine. The product exhibited physical properties and spectral characteristics1 in accord with an authentic sample.