Abstract
Equimolar amounts of N-chlorosuccinimide and sodium iodide in acetone are found to be a convenient source of N-iodosuccinimide using which trans-1,2-iodoacetates and α-iodo carbonyl compounds have been prepared from olefins and enol silyl ethers respectively.
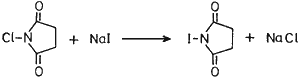
N-Iodosuccinimide (NIS), a convenient source for electrophilic iodination, has been known1 to practising organic chemists for a long time. Although NIS could be prepared by the literature2 procedure from N-silver succinimide and iodine, the former being prepared from succinimide and silver oxide, we sought a newer, cheaper and convenient approach of its preparation. Thus we have found that N-chlorosuccinimide (NCS) upon treatment with sodium iodide in acetone formed NIS and sodium chloride precipitated out. Filtration of NaCl and evaporation of the solvent gave essentially pure NIS which was used as such without further purification (see experimental).
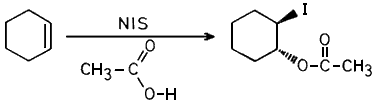
Thus reactions using this NIS were compared with that using commercially obtained NIS. We wish to report that various olefins, when treated with either commercially obtained NIS or the one prepared from NCS/NaI in presence of acetic acid gave almost identical results (see Table I). i.e. both resulted in the formation of trans-1,2-iodoacetates as reported earlier.1b
The above reactions clearly indicate that the new method for NIS preparation could be very useful in view of the fact that NIS could be either in situ generated or prepared prior to its use from NCS and NaI (both being stable under normal laboratory conditions). The general difficulty with NIS, that it may decompose considerably if not stored properly (protection from light and neat is necessary) may be eliminated if the above described simple procedure is adopted.
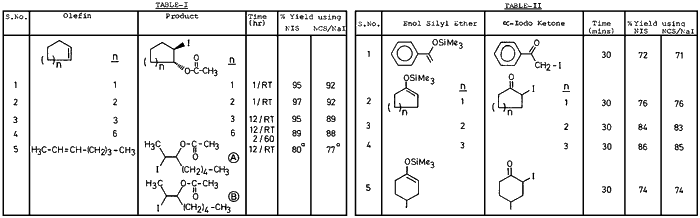
a) Ratio of the products (A) and (B) were found to be 30:70 by spectral analysis
Experimental
1. Preparation of trans-cyclohexane-1,2-iodoacetate
Equimolar amounts (1.83 mmols) of NCS (245 mg) and NaI (274.5 mg) were dissolved separately in dry acetone (2.5 ml) and mixed together at room temperature, The resulting mixture was stirred for 10 mins, excess acetone (10 ml) was added to it and the precipitated NaCl was filtered. Acetone was then removed completely under vacuum to obtain a residue which was suspended in chloroform (5 ml) and further treated with 1.22 mmol (100 mg) of cyclohexene dissolved in chloroform (1 ml) and acetic acid (2 mmol, 0.139 ml). After stirring for 1 hr, at room temperature, the mixture was diluted with ether, washed with 2N sodium carbonate solution (2x10 ml), 5N sodium thiosulfate solution (1x10 ml), water and brine solution and dried over anhydrous sodium sulfate. The solvent was evaporated and the product purified by column chromatography to afford 277 mg (92% of pure cyclohexene-1,2-iodoacetate (characterised by spectral means).

Furthermore, we have also found that NIS either commercially obtained or prepared from NCS and NaI (or used as such without the filtration of NaCl) when treated with enol silyl ethers gave the corresponding (α-iodo carbonyl compounds in excellent yields. The reaction may be considered to proceed via the intermediate A (shown below) which decomposes to produce α-iodo ketone and water miscible succinimide derivative (B).
It is interesting to note that the experimental conditions are mild and the yields are excellent. The data is summarized in Table II. Although various methods are reported in the literature3 for the preparation of α-iodo carbonyl compounds the present method should constitute a useful addition to them.
2. Synthesis of α-iodo cyclohexanone
Equimolar amounts (1.75 mmols) of NCS (234.5 mg) and NaI (262.5 mg) were dissolved separately in dry acetone (2.5 ml) and mixed together at room temperature and stirred for 10 mins. Complete removal of acetone gave a residue which was suspended in anhydrous THF (5 ml) and 1.176 mmol of enol silyl ether of cyclohexanone (200 mg) was added to the mixture maintained at 0°C and stirred for 30 mins. The solution was poured into a mixture of aqueous sodium bicarbonate (10%, 10 ml) and saturated sodium chloride (10 ml), and extracted with petroleum ether (3x10 ml). The organic layers were combined and dried over anhydrous sodium sulphate. Removal of solvent (under vacuum) gave a pale yellow liquid which on purification by column chromatography gave 220 mg (83%) of α-iodocyclohexanone (bp 55°C at 1 mm).
3. Preparation of NIS
Equimolar amounts (1:1 mmols) of NCS (134 mg) and NaI (150 mg) were dissolved separately in dry acetone (2.5 ml) and mixed together. Resulting mixture was stirred for 15 mins, 10 ml of acetone was added to it, and the precipitated sodium chloride was filtered. The NIS obtained after the complete evaporation of solvent (under vacuum) was pure enough for the above described reactions.