Abstract
The conversion of isomyristicin to a Schiff base of myristicinaldehyde is described. The subsequent hydrolysis of this base to the free aldehyde establishes a convenient preparation of this material. Apiolealdehyde is similarly generated from isoapiole, suggesting that this procedure may have general application in the conversion of the natural aromatic ethers of essential oils to the correspondingly substituted benzaldehydes.
In connection with a study of the syntheses of alkaloids related to Anhalonium lewinii, the need arose for appreciable quantities of myristicinaldehyde (4, R = H). Three preparatory routes were considered: the cyclic ether could be prepared from 5-hydroxyvanillin; 2,3-methylenedioxyanisole could be appropriately formylated; or modifications could be made on the chain of naturally available myristicin.
Examples of the first process which employ methylene bromide1 and methylene sulfate2 in the formation of the methylenedioxy ring have been reported. In both instances the yields of myristicinaldehyde were prohibitively poor. Present attempts in this manner employing methylene iodide proved to be equally impractical.
The Vilsmeier reaction, which adds a formyl group to the aromatic ring, has been applied to 2,3-methylenedioxyanisole. The earliest report3 described a 46% yield of product substituted adjacent to the methoxyl group, but subsequently it was observed that an appreciable percentage of substitution also occurred adjacent to the methylenedioxy group4. A careful gas-liquid chromatographic analysis has been performed of the crude reaction mixture. The two isomers described are indeed the principal components, but a third peak of the desired orientation was consistently present. It amounted to only 5% of the total aldehyde content, however, and no experimental variation could be found to increase this yield.
The third route, that which starts with myristicin, appeared to be the only practical synthesis of the title compound. Base-catalyzed isomerization to isomyristicin (1) places the double bond in a position appropriate to an aldehydic end product. KMnO4 has been employed in a number of procedures5 to provide the final aldehyde, but there is concurrent formation of the corresponding benzoic acid. Hydrogen peroxide with a vanadium catalyst6 precludes acid formation but still provides only modest yields. It has only been through ozonolysis that sizable quantities of 4 have been prepared7 but here minor variations in the reaction conditions seriously affect the yields realized7c.
It has been observed in the course of the preparation of a number of β-nitrophenylpropenes from the corresponding olefinic precursors8, that there was invariably produced a small amount of the aromatic aldehyde that would result from cleavage of the propenylbenzene at the double bond. This hydrolysis of the product nitropropene into its theoretical synthetic precursors (benzaldehyde and nitroalkane) was explored as a preparative route to these aldehydes which are otherwise difficult to obtain.
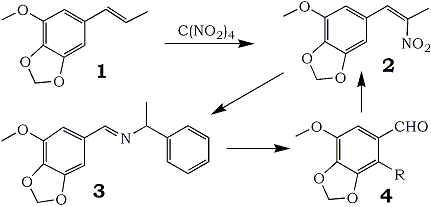
It has been found that this hydrolytic reaction is the principal reaction to occur when an amine that can form a Schiff base (3) is introduced. The completion of the synthetic cycle by the base-catalyzed condensation of the product aldehyde with nitroethane to regenerate the nitropropene (2) both verifies the structure of the aldehyde generated and confirms that the action of tetranitromethane is indeed on the β-carbon of the phenylpropene. Of a number of bases explored as agents in this displacement, primary amines related to benzylamine gave the most consistent results, and of these DL-α-methylbenzylamine appeared to be the best.
The reaction is allowed to occur between stoichiometric amounts of the benzylamine and the nitropropene. If the amine is employed in excess as a solvent a material that corresponds to a 1:1 adduct of these two components is obtained as the principal product.
This procedure has been successfully employed in the preparation of apiolealdehyde (4, R=OCH3) from isoapiole (85%) and appears to be generally applicable to nitropropenes derivable from natural essential oils.
Experimental
1-(3-Methoxy-4,5-methylenedioxyphenyl)-2-nitropropene (2)
A solution of 50 g isomyristicin (1) in 300 ml dry acetone and 24 g pyridine was cooled to 0°C with vigorous stirring. There was then added 54 g of cold tetranitromethane which caused a slight temperature rise (to 5°C) despite the external cooling. The stirring was continued for 2 min at which time the reaction mixture was quenched with a chilled solution of 16.8 g KOH in 300 ml H2O. Stirring was continued while the heat of neutralization dissipated. and the product was then removed by filtration. An additional quantity of the nitropropene (for a total of 50.7 g, 82%) was obtained from the filtrates by extraction with methylene chloride. The low melting point (103°C) was due to a small contamination with myristicinaldehyde which is generated concurrently. Although it cannot interfere with the subsequent reaction below, it can be removed by recrystallization from methanol to yield yellow needles of 2, mp 110°C.
Myristicinaldehyde (4; R = H)
An intimate mixture of 50g 2 and 26g DL-α-methylbenzylamine was heated on the steam bath until solution was effected and the evolution of nitroethane was complete. The intermediate Schiff base 3 was hydrolyzed without isolation by the addition of dilute hydrochloric acid and continued heating. At 60-80°C there was an abrupt solidification of the myristicinaldehyde that had formed and further heating was unnecessary. The aldehyde is removed by filtration of the cooled reaction mixture and could be purified either by crystallization from water, or better by exhaustive extraction of this crude solid product with boiling hexane. The yield thus obtained is 36.9 g (97% of theory and 79% overall from isomyristicin) of a cream-colored solid with mp 128-129°C.
Complete integrity of the ether orientation was established by the reconversion of 4 to 2 with nitroethane as has been described for the trimethoxy counterpart8.
Preparation of the Nitropropene:α-Methylbenzylamine Adduct, C19H22N2O5·xH2O
When an excess of the benzylamine is employed as a solvent, the addition of water at the end of the above-mentioned dissolution resulted in an intensely insoluble product, mp 161°C (decomposition) which is soluble in neither the aqueous nor the organic phase. This white solid, on washing with acetonitrile, showed an analysis that indicated it to be an adduct of one molecule of each of the reactants, with about two molecules of water of hydration.
A more convincing argument for this molecular formula comes from mass spectrometric analysis, which showed a molecular ion at m/e 358 which corresponds to the water-free 1:1 adduct. A daughter ion at m/e 283, corresponding to the loss of the CH3CH2NO2 moiety, is confirmed by an appropriate metastable peak. An independent daughter peak at m/e 237 indicated the loss of α-methylbenzylamine moiety but with the retention of the nitroethane; this corresponds to the production of an ion corresponding to 2. The use of benzylamine led to a similar product (mp 152°C decomposition) and lends weight to the suggested stoichiometry*. Although the microanalytical results indicated some two or three molecules of water of hydration, again the mass spectrum showed only the molecular ion peak corresponding to the anhydrous 1:1 adduct (C18H20N2O5; m/e 344). and both daughter ions corresponding to the loss either of nitroethane (m/e 269) or of benzylamine (m/e 237).
Apiolealdehyde (4, R = OCH3)
Employing the procedure described above for isomyristicin, isoapiole (mp 55–56°C obtained from apiole of Oil of Parsley) was nitrated to 1-(2,5-dimethoxy-3,4 methylenedioxyphenyl)-2-nitropropene, and this intermediate was similarly converted to the aldehyde. This product aldehyde (mp 102–103°C) was obtained in a 75% overall yield from isoapiole.
Formylation of 2,3-Methylenedioxyanisole
The crude reaction product, obtained from the interaction of 2,3-methylenedioxyanisole with the Vilsmeier complex generated from N-methylformanilide and POCl3, was analyzed by gas-liquid chromatography. Complete separation of the three possible benzaldehydes was achieved through the use of an ethylene glycol succinate column (15%, 150×1 cm) at 190°C. The two major peaks emerged at 7.8 min and at 12.0 min, and were readily identified as 2-methoxy-3,4-methylenedioxybenzaldehyde and 4-methoxy-2,3-methylenedioxybenzaldehyde. respectively. The third aldehyde, myristicinaldehyde, occurred as a minor peak at 9.5 min. A characteristic ratio for these materials was 45:9:1, and variation in reaction condition caused marked variation in total yield but little change in this ratio.
* Both of these hydrated adducts showed in their infrared spectra, a salt-like structure involving the N-H bond which indicated an intramolecular interaction with the nitro group.