3.11. Mescaline (β-3,4,5-Trimethoxyphenethylamine), C11H17O3N
Mescaline, the active hallucinatory principle of the "mescal buttons" or "pellote", was isolated by Heffter96 in 1896. Pellote (Anhalonium lewinii Hennings) contains up to 6% mescaline. Reti97,98 observed the presence of mescaline in the Argentine cactus Trichocereus terscheckii, Britton and Rose, which contains 0.2% trichocereine (dimethylmescaline) and 0.05% mescaline. Herrero-Ducloux99 has assumed that one of the bases extracted from Echinocactus gibbosus D.C., (Gymnocalycium gibbosum Pfeiff.) was mescaline.
Microchemical reactions of mescaline have been described by Rosenthaler100, Herrero-Ducloux101, and Bolland102. The free base is a colorless strongly alkaline oil or crystals having mp 35-36¯C (Kindler and Peschke103), bp 180¯C/12mmHg. It is soluble in water, alcohol, and chloroform, but only slightly so in ether. It readily absorbs carbon dioxide from the air forming the solid carbonate. The sulfate (C11H17O5N)2ñH2SO4ñ2H2O is particularly suited for isolation since it is insoluble in alcohol, only slightly soluble in cold water but very soluble in hot water; it forms brilliant prisms, mp 183-186¯C. The hydrochloride forms colorless crystals, mp 184¯C; picrate, mp 222¯C; the aurichloride crystallizes with 1 H2O, orange needles, mp 140-141¯C; platinichloride, straw-yellow needles, mp 187-188¯C; benzoyl derivative, mp 123¯C; m-nitrobenzoyl derivative, mp 161-102¯C; dimethylmescaline methiodide, mp 226-228¯C.
Heffter104-106 determined the empirical formula of mescaline and found that upon oxidation the base yields trimethylgallic acid. Unfortunately, mescaline behaves in methylimino determinations as though it contained an N-methyl group. Heffter107 synthesized 3,4,5-trimethoxybenzylmethylamine and found that it was not identical but isomeric with mescaline.
The irregularity mentioned was confirmed by Spðth3 who, guided by biogenetical considerations, arrived at the correct structure, in spite of the confusing analytical evidence. In Spðth's mescaline synthesis 3,4,5-trimethoxybenzoyl chloride was reduced by the Rosenmund method108 to the corresponding aldehyde, which when condensed with nitromethane yielded ω-nitro-3,4,5-trimethoxystyrene. This was reduced with zinc dust and acetic acid to the corresponding oxime and the latter was further reduced, by sodium amalgam, to β-3,4,5-trimethoxyphenethylamine, i.e., mescaline. Subsequently a number of improved syntheses have been reported.
Slotta and Heller109,110 prepared their starting material, viz., trimethoxyphenylpropionic acid by condensation of the substituted benzaldehyde with malonic acid and reduction of the resulting cinnamic acid. Mescaline was then obtained by Hofmann degradation of the trimethoxyphenylpropionamide. Kindler and Peschke103 synthesized very pure, crystallized mescaline by condensation of 3,4,5-trimethoxybenzaldehyde with potassium cyanide, acetylation, and catalytic reduction to the amine. Slotta and Szyska111,112 improved Spðth's first synthesis, obtaining mescaline directly by the electrolytic reduction of ω-nitrotrimethoxystyrene. Hahn and Wassmuth113,114 started from elemicine and first prepared trimethoxyphenylacetaldehyde by ozonization. The oxime was then reduced to mescaline. Kindler115 and Kindler and Peschke38,103 improved the catalytic reduction of ω-nitrostyrenes to the corresponding phenethylamines. Hahn and Rumpf116 described the preparation of mescaline by reduction of m-nitrotrimethoxystyrene with Adam's catalyst. A review has been published by Jensch117.
Several isomers of mescaline as well as mescaline-like compounds have been synthesized by Jansen118-120; Slotta and Heller110; Slotta and Szyska111,112; Slotta and M■ller121; Grace122; Iwamoto and Hartung123, and Hey124. None of these compounds cause the euphoric state produced by mescaline.
A: Synthesis of Mescaline (Kindler & Peschke)103
i. 3,4,5-Trimethoxyacetylmandelonitrile
Twenty grams 3,4,5-trimethoxybenzaldehyde, prepared according to Slotta and Heller110, is mixed with 40 mL of saturated sodium bisulfite solution. The separated bisulfite compound in a slurry with water is treated with a solution of 9.5g of potassium cyanide in 20 mL water. The resulting nitrile is filtered, washed first with bisulfite solution then with water, and finally dried on a porous plate. The mandelonitrile is acetylated by boiling for 1 hour with 100 mL of acetic anhydride. The excess anhydride is distilled, the residue is dissolved in ether, and the solution washed successively with sodium carbonate solution, with bisulfite solution, and with water. The residue from the dried ether solution distils at 163-165¯C (0.1 mm.); yield, 82% based on the 3,4,5-trimethoxybenzaldehyde.
ii. Mescaline
Twenty-two grams of 3,4,5-trimethoxyacetylmandelonitrile is dissolved in 200 mL glacial acetic acid and the solution dropped into a suspension of 3 g palladium black in 75 mL acetic acid and 5 mL concentrated sulfuric acid (for apparatus see Arch. Pharm. 1931, 74). The introduction is made with agitation, at 18¯C, and under a hydrogen pressure of 2 atm. In 2.5 hours, 95% of the calculated amount of hydrogen is absorbed. An amount of potassium carbonate equivalent to the sulfuric acid is added, the acetic acid is eliminated in vacuo, and the residue dissolved in water. The aqueous solution is washed twice with ether, treated with excess potassium hydroxide, and the separated mescaline taken up in ether. The residue from the ether extract distils at 173¯C (10 mm.) and solidifies to white crystals, mp 35-36¯C.
B: Synthesis of Mescaline (Slotta & Syszka)111
i. 3,4,5-trimethoxybenzoyl Chloride
Five hundred grams of 3,4,5-trimethoxybenzoic acid (cf. Gilman-Blatt, Organic Syntheses, Coll. Vol. 1, p. 537, John Wiley & Sons, New York, 1941) is added to 285 mL of thionyl chloride freshly distilled over linseed oil and the mixture is heated for 2 hours on a water bath. The still-hot mixture is then distilled under reduced pressure from a Claisen flask, avoiding rubber stoppers. There is obtained 510 g (93% of theory) of 3,4,5-trimethoxybenzoyl chloride boiling at 185¯C/18mmHg.
ii. 3,4,5-Trimethoxybenzaldehyde
To a solution of 200 g of 3,4,5-trimethoxybenzoyl chloride in 1000 mL of xylene freshly distilled over sodium, there is added 60g of a 5% palladium-barium sulfate catalyst. The mixture is heated in an oil bath maintained at 150¯C and a vigorous stream of hydrogen is introduced into the boiling solution. The hydrogen should he washed with aqueous permanganate and then dried with sulfuric acid. After 60-80 hours the reaction is complete. The solution is filtered and the aldehyde conveniently isolated as its bisulfite compound. Yield 120g (70.6% of theory), mp 74¯C.
iii. 3,4,5-trimethoxy-beta-nitrostyrene
A solution of 40 mL of nitromethane and 100 g of 3,4,5-trimethoxybenzaldehyde in 200 mL alcohol is cooled to 0¯C and while it is stirred mechanically there is introduced a solution of 45 g pure potassium hydroxide in 45 mL water and 90 mL methanol at the rate of about one drop per second, care being taken that the temperature does not rise. Fifteen minutes after the addition is completed the solution is poured into 500 mL concentrated hydrochloric acid mixed with sufficient ice to assure its presence throughout the slow addition and to maintain a temperature of -10¯C. The precipitated nitrostyrene is separated by filtration and washing and may be purified by recrystallizing from 700 mL alcohol. The pale yellow plates which melt at 120-121¯C are obtained in a yield of approximately 78% of theory.
Figure 1.
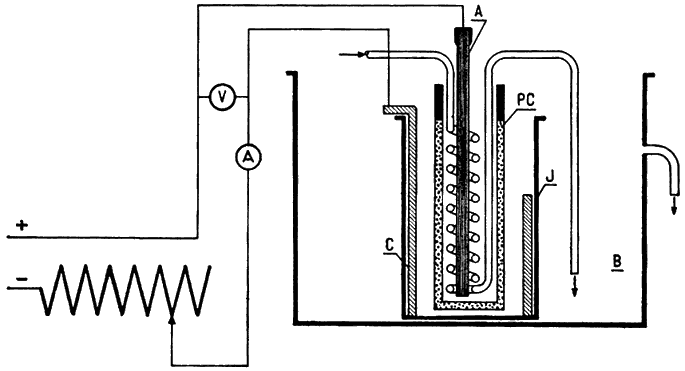
iv. Mescaline
Apparatus (Fig. 1)
A cell of porous porcelain (PC) (external dimensions: 75 x 160) with a glazed rim is placed in a glass jar of 500 mL capacity (J), surrounded by a cooling bath (B). The anode (A) is a load or carbon rod, surrounded by a glass coil; the cooling water flows through the coil and discharges into the cooling bath. The cathode (C) is a sheet of lead (220x90x2 mm), which previous to each experiment is electrolytically coated with lead peroxide, in a bath of dilute sulfuric acid.
Reduction
The cathode liquor consists of a solution of 30 g 3,4,5-trimethoxy-ω-nitrostyrene in 100 mL glacial acetic acid and 100 mL alcohol, to which 50 mL conc. hydrochloric acid has been added. The anodic compartment is filled, to the same level occupied by the catholyte, with a solution of 25 mL conc. sulfuric acid in 175 mL water.
The reduction requires 12 hours, using a current of 5-6 amperes; the cathode current density should be about 3 amperes per square centimeter. The temperature is regulated by the flow of the cooling water and the catholyte should be kept at 20¯C for the first six hours; the temperature is then allowed to rise until it reaches 40¯C at the end of the reduction.
When the reduction is complete, the catholyte is filtered, evaporated in vacuum and the residue taken up in 300 mL water. Unreduced nitrostyrene is extracted successively with ethyl acetate and with ether. The crude mescaline hydrochloride solution in a separatory funnel is then treated with a cold concentrated solution of 100g of sodium hydroxide and the liberated base exhaustively extracted with ether. The somewhat concentrated and dried (potassium carbonate) solution is treated with a stream of dry hydrogen chloride and the separated hydrochloride twice recrystallized from absolute alcohol. The pure mescaline hydrochloride thus obtained in 77% yield forms white leaflets melting at 184¯C.
An improved synthesis of mescaline has been described by Benington and Morin175. 3,4,5-Trimethoxy-β-nitrostyrene176 was reduced with lithium aluminum hydride, using a method described by Ramirez and Burger177. The yield was of 86%.
A new synthesis of mescaline has been recently elaborated by Tsao178. The synthesis is outlined as follows, gallic acid → 3,4,5-trimethoxybenzoic acid → methyl ester of the 3,4,5-trimethoxybenzoic acid → 3,4,5-trimethoxybenzyl alcohol → 3,4,5-trimethoxybenzyl chloride 3,4,5-trimethoxyphenylacetonitrile → mescaline. The reduction of the methyl ester and of the nitrile has been achieved using lithium aluminum hydride.