Synthesis of alpha-Methyltryptamine (IT-290; AMT)by Rhodium[ Back to the Chemistry Archive ] Introduction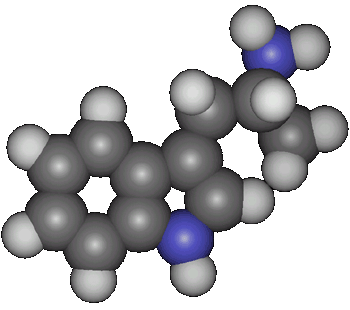
alpha-Methyltryptamine Alternate names: 3-(2-Aminopropyl)indole. Indopan (USSR), IT-290 (d,l form) (Sandoz), IT403 (d form) (Sandoz), U-14,164E (d,l form) (Upjohn), AMT. LD50: 38 mg/kg i.p. in the mouse and 22 mg/kg orally in the rat (Toxicol. Appl. Pharmacol. 4, 547 (1962)).
SynthesesAMT from Indole-3-carboxaldehyde [1]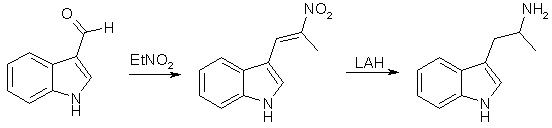
Method A alpha-Methyl-beta-indolenideniumethyl Nitronate (1) With Ammonium Acetate A mixture of 22.0g (0.28 mole) of crystalline ammonium acetate, 6 ml acetic anhydride, and 20 ml of glacial acetic acid was stirred and warmed for approximately 20 min. A mixture of 28.8 g (0.2 mole) of indole-3-aldehyde, 100 ml nitroethane, and 120 ml of glacial acetic acid was added to the solution. When the mixture was brought near reflux, 14.0 g of anhydrous sodium acetate was added. At reflux 20 ml of acetic anhydride was added to the dark solution during 2 h. After 2 h the solution was allowed to cool while 45 ml of water was slowly added. The solid was collected and washed with a solution of 100 ml acetic acid and 45 ml of water. After crystallisation from dilute alcohol, the product weighed 20.2 g (50%) and melted at 190-192řC. The analytical sample melted at 192-193°C. (2) With ammonium phosphate A mixture of 7.2 g (0.05 mole) of indole-3-aldehyde, 7.2 g (0.054 mole) of dibasic ammonium phosphate, 25 ml of nitroethane, and 28 ml of acetic acid was refluxed for three hours, allowed to cool and filtered. After two crystallisations from alcohol, 6.0 grams (60%) of product was obtained. It was identical with that obtained from the experiment with ammonium acetate. alpha-Methyltryptamine Five grams (0.024 moles) of alpha-methyl-beta-indolenideniumethyl nitronate was placed in a drip extractor. A mixture of 5.7 g (0.15 mole) of lithium aluminum hydride and 2000 ml of ether was stirred and refluxed for 4 hours, util all the compound was extracted into the reaxtion mixture. The mixture was decomposed with wet ether, followed by the addition of water and then potassium hydroxide. The suspension was filtered and the filtrate was dried over potassium carbonate and concentrated. The residue was crystallized from ethyl acetate-petroleum ether (bp 60-71°C) to give 2.0 gram (71%), mp 97-100°C. Method B A solution of 10.0g (0.049 mole) of 3-indolyl-beta-methyl-beta-methylethylene [2] in 100 ml of tetrahydrofuran was added dropwise (over 2.5 h) to a mixture of 10.7 g (0.28 mole) of lithium aluminum hydride in 100 ml of tetrahydrofuran. The mixture was gradually heated to reflux during the addition. After the addition was complete the mixture was refluxed for 2 h and allowed to stand overnight. A solution of 20 ml water and 60 ml of tetrahydrofuran was slowly added until the excess lithium aluminum hydride was destroyed, followed by 10 ml of concd sodium hydroxide. Ether (150 ml) was then added and the mixture was rapidly stirred until no solids remained on the sides of the flask. The mixture was filtered and the solid was washed with 150 ml of ether. The ether solutions was combined, dried over potassium carbonate, and concentrated to yield 9.2 of crude amine. The amine was dissolved in 110 ml of methanol and 5.5 ml of acetic acid was added. The solution was concentrated to dryness under reduced pressure and the residue was dissolved in 110 ml of hot ethyl acetate upon cooling 8.2 g (73.2%) of alpha-methyltryptamine acetate precipitated. After drying the salt melted at 143-144°C. AMT and AET from Gramine [2]In this article, nitromethane, nitroethane, 1-nitropropane and ethyl nitroacetate are alkylated with gramine. The nitroethane derivative gives alpha-methyltryptamine on reduction. The procedures are described for 1-nitropropane, but is applicable on the whole series of nitro compounds. (1) Alkylations of Nitro Compounds with Gramine The reaction with 1-nitropropane was carried out by refluxing a solution of 10 grams of gramine in 50 ml of redistilled nitropropane in the presence of 2.6g of solid sodium hydroxide. Nitrogen was passed through the system before heating was begun. Dimethylamine was evolved copiously, and refluxing was continued six to eight hours, or until the evolution of the amine had nearly ceased. The mixture was then cooled and acidified with 50 ml 10% aqueous acetic acid. The resulting mixture was diluted with 200ml of ether and washed four times with 75 ml portions of water. The solution was then shaken with Norit and filtered. The solvents were removed by distillation under reduced pressure at room temperature, leaving a viscous brown oil; which was distilled at 0.2 mmHg, bp 157°C. The condensation product could with difficulty be crystallized, mp 90-91°C. (2) Hydrogenations of the Nitro Compounds A solution of 15g of the condensation product from 1-nitropropane in 150 ml of ethanol was refluxed for 15 minutes with 1/4 teaspoonful of Raney Nickel catalyst. The nickel was filtered from the cooled solution, which was then placed in a hydrogenation bottle with 0.2 grams of Adams catalyst. The hydrogenation was conducted at room temperature and under an initial pressure of about 50 lb. and it was allowed to proceed for about thirty-six hours. After filtration from the catalyst the solution was concentrated and the residue dissolves in 200 ml of benzene. The basic material recovered by the extraction of the benzene solution with three 150 ml portions of 2 N hydrochloric acid which were added to 500 ml of 2 N sodium hydroxide weighed 8 g and melted 93-95°C. The analythical sample (mp 101-102°C) was prepared by crystallization from benzene. Other preparations of amines were carried out in essentially the same manner. Using this method, the yield of alpha-ethyltryptamine approaches 95% but in the case of alpha-methyltryptamine, this only amounts to around 10%. AMT From Indole [3]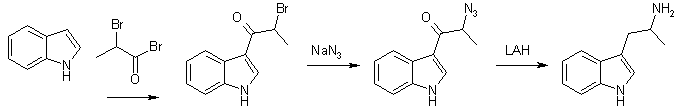
3-(2-Bromopropionyl)indole. (A) alpha-bromo-propionyl bromide (21.6g, 0.1 mol) was added dropwise during one hour to a well-stirred solution of indole (11.7g, 0.1 mol) and pyridine (8.1 ml, 0.1 mol) in dioxan (100 ml) at 60°C. The mixture was stirred for another hour, cooled and poured into water (500 ml). The oil formed was separated and dissolved in MeOH. On standing (1-24h) crystals separated. Recrystallization from acetonitrile gave 3-(2-bromopropionyl)indole free from the 1-isomer, yield 15.2g (60%), mp 210-212°C. 3-(2-Bromopropionyl)-indole. (B) As in method A, but the dioxan (100ml) was replaced with toluene (300ml), yield 18.4g (72%), mp 210-212°C. 3-(2-Azidopropionyl)-indole 3-(2-Bromopropionyl)indole (8.4g) was added in 10 portions during 15 minutes to a well-stirred suspension of sodium azide (3.3g) in DMSO (40 ml) at 25°C. After completed addition the mixture was stirred for two hours, and then poured into water (400 ml) and extracted with ether. The ether phase was washed with H2O, dried and evaporated. The residue recrystallized from ether/petroleum ether gave 5.2g (73%), mp 125-126°C. 3-(2-aminopropyl)indole (AMT) 3-(2-Azidopropionyl)indole (3.5g) in dry THF (50 ml) was added to a well-stirred mixture of LAH (4.0g) in dry THF (100ml). After a reflux period (23h), the excess LAH was destroyed by addition of KOH (2M, 5ml). The mixture was filtered and the solid inorganic phase carefully washed with ether. The ether phase was dried and evaporated. The residue recrystallized from hexane/EtOAc gave 2.1g (74%), mp 100-102°C. (+) And (-) AMT From Tryptophan [4]Enantiomerically pure alpha-methyltryptamine can be made through reduction of the ethyl esters of D- and L-tryptophan, respectively. (+)-AMT is approximately four times as potent a CNS stimulant as (-)-AMT. R(+)-2-Amino-3-(3-indolyl)propanol One g. (3.58 mmoles) of D-tryptophan ethyl ester hydrochloride was added in portions to a stirred suspension of 800 mg (21 mmoles) of lithium aluminum hydride in 15 ml. of dry tetrahydrofuran at room temperature. After stirring for 30 minutes, the complex was decomposed by dropwise addition of 2N sodium hydroxide. The solids were filtered and shaken with 50 ml. of 2N sodium hydroxide and 200 ml. of chloroform in a separatory funnel. The organic layer was separated, combined with the original filtrate and dried (magnesium sulfate). The drying agent was removed by filtration and the filtrate concentrated at reduced pressure. The syrupy residue was crystallized from ethyl acetate/ hexane, yield 450 mg (66%). R(+)-N-(Benzyloxycarbonyl)-2-amino-3-(3-indolyl)-propanol R(+)-2-Amino-3-(3-indolyl)propanol (1.32 g., 6.95 mmoles) was dissolved in a mixture of 30 ml. of water and 30 ml. of acetone. Sodium carbonate (1.27 g., 12 mmoles) was added and to the stirred, cooled mixture (ice) was added dropwise 1.0 ml. (7.0 mmoles) of benzyl chloroformate. After the addition the cooling bath was removed and the reaction stirred at room temperature for 1.5 hours. The reaction mixture was acidified (to pH 2) with concentrated hydrochloric acid and diluted with 100 ml. of water. The aqueous mixture was extracted with 2x150 ml of ethyl acetate, the organic solution washed with 100 ml. of saturated aqueous sodium chloride and dried (magnesium sulfate). Filtration of the drying agent and concentration in vacuo left a syrupy residue which was crystallized from chloroform/hexane to give 1.7 g. (75%). R(+)-N-(Benzyloxycarbonyl)-2-amino-3-(3-indolyl)propanol p-Toluenesulfonate R(+)-N-(Benzyloxycarbonyl)-2-amino-3-(3-indolyl)-propanol (350 mg., 1.08 mmoles) was dissolved in 10 ml. of dry pyridine and 310 mg (1.62 mmoles) of p-toluenesulfonyl chloride was added. The reaction was stored at room temperature for 18 hours and the solvent distilled under reduced pressure. The residue was partitioned between 200 ml of ethyl acetate and 50 ml. of saturated aqueous sodium chloride. The organic layer was washed with 50 ml. of water and dried (magnesium sulfate). Filtration and concentration in vacuo left a foamy residue. Pure product was isolated by preparative TLC using 10% acetone in benzene, yield 400 mg (77%). This compound was unstable at room temperature but could be stored for several weeks at -15°C. S(+)-3-(2-Aminopropyl)indole p-Toluenesulfonate R(+)-N-(Benzyloxycarbonyl)-2-amino-3-(3-indolyl)propanol p-Toluenesulfonate (400 mg., 0.84 mmole) was dissolved in 25 mL of absolute ethanol and 100 mg. of 10% palladium on charcoal catalyst added. The reaction mixture was shaken under 3 atmospheres of hydrogen for one hour. The catalyst was filtered (Celite) and the filtrate concentrated under reduced pressure. The residual oil was taken up in 6 ml. of hot chloroform and cooled to room temperature. The precipitate was filtered and dried in vacuo. It was recrystallized from methanol/ether, yield 240 mg (82%). S(+)-3-(2-Aminopropyl)-indole (S(+)-alpha-methyl-tryptamine) S(+)-3-(2-Aminopropyl)indole p-Toluenesulfonate (100 mg., 0.289 mmole) was stirred in 10 ml. Of 2N sodium hydroxide for 5 minutes. The oily product was extracted with 2x50 ml. of ethyl acetate and the organic solution was dried (magnesium sulfate), filtered and concentrated under reduced pressure. The syrupy residue was crystallized from ethyl acetate/hexane, yield 35 mg. (69%). AMT from Indole and 2-nitropropene [5]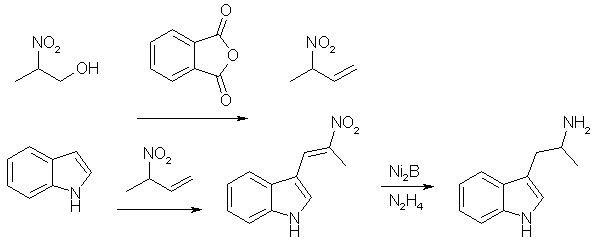
2-Nitropropene In a round-bottomed flask equipped with a vigreaux column and distilling head, were placed 2-nitro-1-propanol and 2.0 molar equivalents of phtalic anhydride. The reactants were heated (oil bath) until a homogenous solution was formed, and the nitro-olefin was distilled over at reduced pressure (41°C at 76 mmHg). The yield was 94% of a pale-green liquid. A small amount of water which was co-distilled was removed with a pipet, and the material was stored as a 10% w/v solution in benzene over anhydrous CaCl2. Nickel Boride The catalyst was prepared just prior to use by the dropwise addition of 2-molar equiv. of sodium borohydride of 1.0 M solution in 0.1 M aqueous sodium hydroxide) to a stirred 0.1 M solution of nickel(II)acetate tetrahydrate in a large beaker. After gas evolution had completely ceased the aqueous solution was decanted, the black granular nickel boride was resuspended in distilled water, then again decanted. After several quick washings with water, the catalyst was washed twice with ethanol and transferred to a round-bottomed flask for use in the reduction. 1-(Indol-3-yl)-2-Nitropropane To a stirred 0.5 M benzene solution of indole was added 2 equiv. of 2-nitropropene as a 10% benzene solution. The reaction mixture was heated at reflux under a nitrogen atmosphere until TLC analysis (dichloromethane/silica gel) indicated that all indole was consumed. The dark reaction mixture was cooled to room temperature and quickly passed through a short silica gel column which was washed with 1:1 toluene/hexane until product recovery was complete. Solvent removal under reduced pressure afforded 1-(Indol-3-yl)-2-nitropropane as an amber oil (81% yield), sufficiently pure (by TLC) to be carried on to the next step without further purification. alpha-Methyl-tryptamine To a stirred suspension of nickel boride in isopropyl alcohol prepared from 5.0 molar equiv of nickel acetate was added the 1-(Indol-3-yl)-2-nitropropane to form a 0.1 M solution. This was heated to reflux and a solution of 10.0 equiv. of hydrazine hydrate in isopropyl alcohol was added dropwise at such a rate that the evolution of gas did not cause excessive forming. After gas evolution was complete, reflux was continued for an additional 30 min, at which time the TLC analysis indicated that the reaction was complete. The mixture was cooled to room temperature and filtered through a pad of Celite, and the filtrate was concentrated under reduced pressure. The residue was dissolved in dichloromethane and extracted with several portions of 10% acetic acid. The combined aqueous extracts were basified with ammonium hydroxide and the product was extracted into several portions of dichloromethane. The combined extracts were dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The 1-(Indol-3-yl)-2-aminopropane (alpha-methyltryptamine) was then recrystallized from diethyl ether. Yield 69% of theory, mp 101-102°C. References [1] R. V. Heinzelman, Synthesis of alpha-Alkyltryptamines, J. Org. Chem., Vol 25, p 1548-1558 (1960)
| |||||||||||||||||||||