Piperonal, or (3,4-methylenedioxybenzaldehyde) (1), an important fragrance used as a soap additive and a synthetic precursor of Dopa, has been industrially prepared by ozonolysis1 or chromic acid oxidation2 of isosafrole 2. Because of the environmental pollution associated with chromium species and the requirement of a large quantity of electricity and a carefully controlled reaction temperature (0-5°C) for ozonolysis, these demerits have prompted us to develop a nonpolluting and more economical process. Recently, several attempts employing ruthenium tetroxide oxidation combined with m-periodate3 and air-oxidation under γ-ray irradiation4 have appeared.
On the other hand, an electrochemical oxidation would be a promising method for a piperonal synthesis from isosafrole because an electrooxidative bond cleavage of carbon-carbon double bonds is possible in principle. For example, an electrochemical conversion of cyclic enol acetates to keto esters in a MeOH-AcOH-LiClO4 system has been achieved in satisfactory yield.5 However, the direct bond cleavage method leads in some cases to overoxidation of the product aldehyde to give the corresponding carboxylic acid, as observed in the conversion of methyl eugenol to 3,4-dimethoxybenzoic acid.6 A plausible reaction pathway for the electrochemical carbon-carbon bond cleavage would be initial oxygenation of the double bond to the corresponding glycol or a derivative followed by C-C bond cleavage. We describe a two-step electrochemical procedure that leads to a highly selective preparation of 1 from 2 via 3a.

Epoxidation of olefins is efficiently achieved by the halide ion mediated electrochemical oxidation in which bromide ion is found to be the most useful.7 Thus, 2 was subjected to the electrooxidation in MeCN-H2O (7:2) containing 1.5-2.0 equiv of sodium bromide at room temperature. Platinum foils were employed as electrodes, and a constant current was passed (20 mA, 2.83 F/mol) in an undivided cell. The products were epoxide 4 (71% ) and glycol 3a (23%) after chromatography. Unlike the electrochemical epoxidation of isoprenoids, where the use of 0.1-1.0 equiv of sodium bromide was suitable to suppress the formation of the corresponding dibromide,7 the use of more than 1 equiv of sodium bromide was required to provide 3a and 4 in good yield. Dibromide 3c was found to be spontaneously hydrolyzed to give 3b, which collapsed to the epoxide 4 under the electrolysis conditions. Epoxide 4 is unstable under the reaction conditions and suffers partial hydrolysis resulting in the formation of 3a.8 After electrolysis, 1% aqueous sulfuric acid was added to the reaction mixture, which was stirred for 1 h to give 3a (98%) as a diastereomeric mixture (25:75 by NMR). The glycol 3a was subjected to acid-catalyzed dehydration (p-TsOH- benzene), affording ketone 5 (84%), which is a precursor of methyl Dopa.8,9
The efficiency of the epoxidation is dependent upon the pH of the reaction solution. In a MeCN-H2O-NaBr system, the solution at the end of electrolysis was alkaline. On addition of a small amount of an acid, a mixture of 4, 3a, and the corresponding bromohydrin 3b was obtained. Under more acidic conditions such as MeCN-H2O-NaBr-H2SO4, 3b was obtained quantitatively. The nature of the halide ion is also important in the product selectivity. In contrast to the successful results obtained with bromide ion, the use of chloride or iodide ions resulted in a poor yield of 3a (39% for NaCl and 43% for NaI).
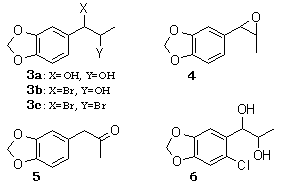
The electrochemical carbon-carbon bond cleavage of glycols has been examined in MeOH-Et4NOTs for cyclic glycols10 and glycol monomethyl ethers11 and in MeOH-NaOH for tetraphenylethylene glycol.12 The related α-hydroxy ketones can be cleaved in MeOH-LiClO4.13 At first, 3a was subjected to electrooxidation both in MeOH-Et4NOTs and MeOH-LiClO4, affording 1 in 66% and 44% yields, respectively. When the halide ion promoted electrooxidation was examined, sodium bromide in MeCN-H2O (9:1) provided a satisfactory yield (86%) of 1, while chloride ion gave good to moderate yields in aqueous acetonitrile [Ca(ClO)2 (0.1 equiv) 85%; CaCl2 (0.2 equiv) 71%; NaCl (0.3 equiv) 61%]. Interestingly, a two-layer system consisting benzene and water (6:4) including NaHCO3 or NaOH greatly enhanced the desired bond cleavage. In aqueous NaHCO3 (0.5 equiv to 3a)-benzene the yield of 1 was 62%, and 15% of the starting material was recovered after 6 F/mol when the reaction solution was found to be acidic. Also the yield increased with an increase of the NaHCO3 concentration 1% of 1 with NaHCO3 (equiv): 62% (0.5), 88% (1.9), 90% (3.7)]. Meanwhile, the use of an appropriate concentration of NaOH led to the quantitative formation of 1 [quantitative (1.0 equiv), 96% (2.0 equiv), 91 % (4.0 equiv)]. In contrast, upon electrolysis with NaBr or LiClO4 instead of the base, the desired reaction proceeded very slowly (about 50% of 3a was recovered) and the yield of 1 was poor (16-19%) after 6 F/mol. In a benzene-water system 3a dissolves in the aqueous phase and the product 1 exclusively in the organic phase. Hence, the product 1 migrates into the benzene layer so that overoxidation of 1 can be avoided.
| Oxidations of Isosafrole Glycol 3a to Piperonal 1 | |||||
| Oxidanta | Solvent | Temp | Time | Yield | |
| Electrolysis | PhH - H2Ob | 65°C |
99% |
||
| NaIO4 (1.5 eq) | MeOH - H2Oc | 0-5°C |
30 min |
94% |
|
| CAN (2 eq) | AcOH - H2Od | 25°C |
30 min |
93% |
|
| Ca(OCl)2 | PhH - H2Oe | 65°C |
3 h |
100% |
|
- (a) Molar equivalents to 3a
- (b) Benzene/Water 6:4
- (c) Methanol/Water4:3
- (d) Acetic Acid/Water 1:2
- (e) Benzene/Water 2:1
A comparison of the electrochemical and the conventional chemical oxidation of 3a is summarized in Table I. Among the oxidants employed, sodium periodate, ceric ammonium nitrate (CAN), and calcium hypochlorite provided satisfactory yields. However, the use of an excess of expensive reagents such as sodium periodate and CAN is disadvantageous for manufacturing piperonal. On oxidizing 3a with calcium hypochlorite in MeCN-H2O-AcOH, as reported by Nwaukwa and Keehn14 for several simple glycols, chlorination on the aromatic ring of 3a took place predominantly, leading to 6. In contrast, oxidation of 3a in a two-layer system of benzene-water provided 1 quantitatively when 2 equiv of hypochlorite were used. Electrooxidation of 3a in a benzene-water system including a small amount of sodium hydroxide also afforded 1 quantitatively. The electrolysis procedure is simple in operation and economical with respect to the reagents employed, and furthermore it requires no disposal of by-products (e.g., waste Ca compounds).
Experimental:
Electrolysis of Isosafrole (2)
Preparation of Isosafrole Glycol (3a).
A mixture of Isosafrole (2) (100 mg, 0.62 mmol) and NaBr (192 mg, 1.9 mmol) dissolved in MeCN (7 mL) and H2O (3 mL) was electrolyzed in a beaker-type undivided cell (3 cm in diameter and 10 cm in height). A constant current (20 mA, 2.83 F/mol) was passed for 140 min by using platinum foils (2 x 1.5 cm2) as electrodes and a Metronix Model 543B DC power supply. After the electrolysis at room temperature, 0.5 mL of 1% aqueous H2SO4 was added to the mixture, which was stirred for 1 h and neutralized with aqueous NaHCO3. After evaporation of solvents under reduced pressure, the organic substances were extracted with ethyl acetate. The extracts were washed with brine, dried (Na2SO4), and concentrated in vacuo to give a colorless oil, which was chromatographed (Mallinckrodt Silica CC-7 Special), affording Isosafrole Glycol (3a) (116 mg, 98%) as colorless crystals8 whose spectral data were identical with those reported. In an another experiment, after the electrolysis as mentioned above, the mixture was concentrated in vacuo and the organic substances were extracted with ethyl acetate. The usual workup and chromatography provided Isosafrole Glycol (3a) (28 mg, 23%) and 4 (78 mg, 71 %). The structure of 4 was identified spectroscopically by comparison with IR and 1H NMR spectra of the authentic sample prepared by mCPBA oxidation of Isosafrole (2)9.
2-Bromo-1-[3,4-(methylenedioxy)phenyl]-1-propanol (3b).
A mixture of 2 (100 mg) and NaBr (96 mg) dissolved in MeCN (7 mL)-1% aqueous H2SO4 (3 mL) was electrolyzed (20 mA for 125 min, 2.5 F/mol) in a similar manner as described above, affording Isosafrole Glycol (3a)9a (160 mg, quantitative yield) as a colorless oil: bp 110-112°C (0.02 mmHg).
Electrolysis of Isosafrole Glycol (3a)
Preparation of Piperonal (1).
Isosafrole Glycol (3a) (100 mg, 0.51 mmol) dissolved in benzene (6 mL) and 0.5% aqueous NaOH (4 mL) was electrolyzed at 65°C (18 mA for 3 h, 4 F/mol) in a similar manner as described above, affording 1 (76 mg, 99%) as colorless crystals whose spectral and TLC data were consistent with those of an authentic sample.
Oxidation of Isosafrole Glycol with NaIO4
A solution of NaIO4 (82 mg, 0.38 mmol) was added to Isosafrole Glycol (3a) (50 mg, 0.26 mmol) dissolved in MeOH (8 mL) at 0-5°C. The mixture was stirred at the temperature for 30 min. The usual workup provided 1 (36 mg, 94 % ).
Oxidation of Isosafrole Glycol with CAN
Into a solution of Isosafrole Glycol (3a) (100 mg, 0.51 mmol) dissolved in AcOH-H2O (1:2,3 mL) was added CAN (587 mg, 1.07 mmol) dissolved in AcOH-H2O (1:2,12 mL). The mixture was stirred at room temperature for 30 min. The usual workup provided 1 (71 mg, 93% ).
Oxidation of Isosafrole Glycol with Ca(ClO)2
A suspension of Ca(ClO)2 (61 mg, 0.26 mmol) in benzene (2 mL)-H2O (1 mL) was added to a solution of Isosafrole Glycol (3a) (100 mg, 0.51 mmol) dissolved in benzene (4 mL)-H2O (2 mL). After vigorous stirring at 65 °C for 1 h, Ca(ClO)2 (183 mg, 0.77 mmol) suspended in benzene (2 mL)-H2O (1 mL) was added again and the mixture was stirred for additional 2 h. After adding AcOEt and centrifuging a precipitate, the organic substances were extracted with AcOEt and the usual workup provided 1 (75 mg, 98%).
1-[2-Chloro-4,5-(methylenedioxy)phenyl]propane-1,2-diol (6)
A suspension of Ca(ClO)2 (60% purity, 92 mg, 0.39 mmol) in AcOH (0.12 mL)-H2O (1.2 mL) was added drop wise to a solution of Isosafrole Glycol (3a) (50 mg, 0.26 mmol) dissolved in MeCN (2 mL)-CH2Cl2 (1 mL). The reaction mixture was stirred at room temperature for 1 h. Ether extraction followed by usual workup and chromatography provided 6 (50 mg, 85%) as colorless crystals: mp 85-86°C.
3,4-Methylenedioxyphenyl-2-propanone (MDP2P, 5)
A solution of Isosafrole Glycol (3a) (100 mg, 0.5 mmol) and p-TsOH (200 mg) dissolved in a distilled benzene (20 mL) was refluxed for 20 min. The usual workup gave MDP2P (5) (75 mg, 84%)8,9.