The main use of this method is to convert benzyl halides to benzaldehydes and 1-phenyl-2-halopropanes to phenyl-2-propanones, as for example the preparation of MDP2P in 90% yield from bromosafrole.
A reaction between 2-nitropropane and benzylidene-bis-dimethylamine gave acetoxime and N.N-dimethylbenzamide-Benzylidene-bis-piperidine and 2- nitropropane gave acetoxime and benzoylpiperidine. The reactions appear to proceed by formation of aminobenzyl 2-propanenitronates which disproportionate into acetoxime and benzamides, thus establishing a relationship with the reaction of halides with sodium 2-propanenitronate to form esters with subsequent disproportionation. The latter reaction has been found to go well in wet solvents and even in water. The reaction has been extended to synthesize the aliphatic aldehydes, undecanal and dodecanal, as well as 3,4-methylenedioxyphenylacetone and cyclohexadione.
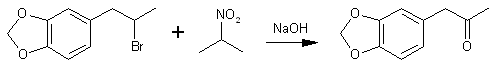
Among a number of routes that could conceivably lead to a phenyl-substituted α-butylamine was a Mannich reaction involving benzaldehyde, dimethylamine and 2-nitropropane. When this reaction was attempted, no Mannich base formed. The products of the reaction were acetoxime and N,N-dimethylbenzamide. The same products, plus dimethylamine, resulted from an attempted abbreviated Mannich reaction1 with preformed benzylidene-bis-dimethylamine and 2-nitropropane. The expected reaction, forming Mannich base, should have been carbon alkylation of the active hydrogen compound, 2-nitropropane, by the cation 1 resulting from the interaction of benzaldehyde and dimethylamine.
The unexpected formation of acetoxime and dimethylbenzamide is explicable only if the two result from the disproportionation of the ester, α-dimethylaminobenzyl 2-propanenitronate, arising from oxygen alkylation of 2-propanenitronic acid by the cation. Although this reaction has not been reported previously, it may be classified among a number of reported reactions2 of halides and salts of nitronic acids giving rise to carbonyl compounds corresponding to the halides and oximes corresponding to the nitronic acids. Earlier examples of similar reactions involve mononitroparaffins other than 2-nitropropane. There can be little doubt that these reactions proceed through the disproportionation of an intermediate nitronic ester into an oxime and a carbonyl compound. The major side-reaction appears to be carbon-alkylation when alkylating with highly activated halogen compounds.
A reaction converting an aldehyde to an amide, while a fascinating facet, seems of limited interest. The same reaction was used to convert benzylidene-bis-piperidine to benzoylpiperidine, but failed with an aliphatic aldehyde, n-hexaldehyde. Heating n-hexaldehyde, dimethylamine and 2-nitropropane led to a partial recovery of starting materials and a 61% yield of the Knoevenagel product, 2-hexylidenehexaldehyde. The formation of the latter product rather than the nitronic ester (and disproportionation products) was probably favored by both the temperature and basicity. The possibility of the Knoevenagel reaction, when catalyzed by secondary amines, proceeding via the bis-amine has been mentioned previously.1 A similar reaction using the preformed bis-dimethylamine from 3,5,5-trimethylhexaldehyde was contemplated, but it was abandoned when only the metholamine, N-(1-hydroxy-3,5,5-trimethylhexyl)-dimethylamine, was obtained from the attempted preparation of bis-dimethylamine.
The two successful examples of amide preparation seem to be dependent on temperature, requiring some forcing. On refluxing benzaldehyde, aqueous dimethylamine and 2-nitropropane in ethanol for two hours, the yield of N,N-dimethylbenzamide was 37%. Benzylidene-bis-dimethylamine and 2-nitropropane without solvent at 120-130°, produced a 94% yield. That the difference is due to temperature and not to the use of preformed bis-amine was shown clearly by the benzoylpiperidine synthesis. Heating benzylidene-bis-piperidine and 2-nitropropane in refluxing ethanol for four hours led to no benzoylpiperidine, 93% of the benzylidene-bis-piperidine being recovered. On dispensing with solvent and heating at 130-140°, benzoylpiperidine was obtained in 54% yield.
The preparation of dimethylbenzamide in aqueous alcohol suggested that the more general reaction, converting halides to carbonyl compounds, might not necessarily require metallic sodium and anhydrous alcohol, and might be a preparative reaction for aliphatic carbonyl compounds under milder conditions. The formation and disproportionation of 2-propanenitronic esters have been found to take place smoothly and with good yields for monocarbonyl compounds. It is not necessary to use metallic sodium and anhydrous alcohol. The reactions go well in 95% ethanol using salts formed in the solvent from 2-nitropropane and sodium or potassium hydroxide before adding the halide. By this procedure 3,4-methylenedioxyphenylacetone, undecanal and dodecanal were obtained in good yield. It has been reported3 that aliphatic aldehydes are not obtained under anhydrous conditions.
To test the feasibility of using water without alcohol, a suspension of benzyl chloride was heated and stirred in an aqueous solution of potassium 2-propanenitronate, giving a 49% yield of benzaldehyde. The possibility that the aqueous conditions might operate against a carbon-alkylation side-reaction had to be discarded when para-nitrobenzyl chloride was treated with an aqueous solution of sodium 2-propanenitronate and found to give a 59% yield of the carbon-alkylation product, 2-methyl 2-nitro-1-(para-nitrophenyl)-propane, and no para-nitrobenzaldehyde.
The presence of acetoxime in water-insoluble aldehydes or ketones presents no problem since it is readily removable from ether solution by water. In some preliminary experiments, last traces of acetoxime appeared as crystals in the condenser from which it was sublimed into the trap by using warm water in the condenser. Acetoxime presents a more serious problem when the other product is also water soluble, e.g., succindialdehyde and 1,2-cyclohexadione. Preparation and separation of a derivative was considered inadvisable because of the amounts involved, and distillation of the mixtures was attempted. Distillation was only moderately successful for cyclohexadione, because decomposition during the prolonged heating held the yield down to 30%. In the attempted preparation of succindialdehyde from 1,4-dichlorobutane, decomposition during distillation was more rapid, and no product could be obtained from the residual tar after removal of the acetoxime fraction. This latter case and other polyfunctional possibilities require a more searching investigation.
Both chloro and bromo compounds have been converted successfully to carbonyl compounds, usually at reflux temperatures. Activated halogen as in 2-chlorocyclohexanone reacts vigorously, and it is advisable to add the reagent slowly with cooling4. Final refluxing is necessary, both for completion of the metathesis and disproportionation of the nitronic ester.
Experimental5
Benzylidene-bis-dimethylamine
Benzaldehyde (106 g., 1 mole) was poured into a 1-liter flask containing 400 g. of 25% aqueous dimethylamine. The mixture was swirled occasionally during 10 minutes warming on the steam-cone. After cooling, the aqueous layer was saturated with potassium carbonate and the upper layer separated with the aid of 100 ml. of benzene. The benzene layer was dried over potassium carbonate, the benzene removed under reduced pressure and the residue distilled. The product was collected at 57-60°C/(0.9mmHg, 143 g. (80%).
N,N-Dimethylbenzamide
Benzylidene-bis-dimethylamine (44.5 g., 0.25 mole) was heated in a 200-ml. flask, fitted with a dropping funnel and reflux condenser with drying tube, in an oil-bath at 120-130°C. 2-Nitropropane (22.2 g., 0.25 mole) was added dropwise in the course of 2.5 hours with continuous evolution of dimethylamine. Heating was continued one additional hour and then the light amber reaction mixture was distilled. The forerun was collected at 35-70°C/20mmHg, and the dimethylbenzamide at 94-96°C/0.5mmHg, 35g (94%).
The forerun was redistilled, giving white crystals in the receiver and condenser, at 55°C/20mmHg. On recrystallization from heptane, it melted at 62-63°C, no depression of mp on mixing with an authentic sample of acetoxime.
Redistillation of the dimethylbenzamide gave bp 266°C, mp 38°C. Because the material was water insoluble6 and the mp low, it was not analyzed.
A portion was hydrolyzed with 20% sulfuric acid, precipitating benzoic acid, m.p. and mixed m.p. 121-122°, after recrystallization from water. The hydrolysis filtrate was made alkaline and distilled, trapping the dimethylamine distillate in ice-water. Addition of phenyl isothiocyanate to the distillate precipitated N,N-dimethyl-N'-phenylthiourea, m.p. 133-135° after recrystallization from ethanol.
Another preparation of N,N-dimethylbenzamide was made by mixing 0.25 mole each of benzaldehyde (26.5 g.), 2-nitropropane (22.2 g.) and 25% aqueous dimethylamine (45 g.), with 50 ml. of ethanol. The reaction was heated under reflux for two hours, cooled, and extracted with benzene (three 50-ml. portions). The solvents were removed under reduced pressure and the residue distilled, obtaining 12.5 g. of benzaldehyde at 40-45° (0.8 mm.), phenylhydrazone m.p. 158°C,7 and 14.0 g. (37%) of N,N-dimethylbenzamide at 100-104° (0.8 mm.).
Benzoylpiperidine
2-Nitropropane (17.8 g., 0.2 mole) and 25.8 g. (0.1 mole) of benzylidene-bis-piperidine8 were mixed in a 100-mI. flask fitted with a reflux condenser. The flask was heated in an oil-bath at 130-140° for three hours. After cooling, the dark oil was poured into 200 ml. of 2.5 N hydrochloric acid and shaken vigorously. The dark heavy layer was taken up in ether, washed with water to remove acetoxime, dried over magnesium sulfate and distilled. After removal of ether and 2-nitropropane, there was obtained 2 g. of benzaldehyde at 46° (0.8 mm.) and 10.2 g. (54%) of benzoylpiperidine at 136-139° (0.8 mm.) as a pale lemon oil which solidified to a white solid, m.p. 46-48°, no depression of m.p. on mixing with an authentic sample.
2-Hexylidenehexaldehyde
This compound was the product of an unsuccessful attempt to prepare N,N-dimethylcaproamide carried out in the following manner, n-Hexaldehyde (50 g., 0.5 mole) was added to 110 g. of 25% aqueous dimethylamine (1.1 moles) and heated for five minutes on the steam-cone. The mixture was cooled, saturated with potassium carbonate, and the upper phase transferred to a 500-ml. flask fitted with a dropping funnel and a reflux condenser. It was heated on the steam-cone while 44.5 g. (0.5 mole) of 2-nitropropane was added drop-wise in the course of two hours, and heating continued for one more hour. After cooling, the mixture was taken up in 50 ml. of ether and washed with 100 ml. of N hydrochloric acid and three 50-ml. portions of water. After drying over magnesium sulfate, ether, unreacted alpha-hexaldehyde and 2-nitropropane were removed at 30 mm. The 2-hexylidene-hexaldehyde, 28 g. (61%), distilled at 80-82° (0.8 mm.). It was redistilled at 79° (0.7 mm.), with virtually no change in constants, nD 1.4566, dt 0.8438, MD 58.80 (calcd. 59.06). The 2,4-dinitrophenylhydrazone melted at 132-133° (from ethanol).
Aqueous dimethylamine (200 g. of 25%, 1.1 moles) in a 500-ml. flask was cooled in an ice-bath and 71 g. (0.5 mole) of 3,5,5-trimethylhexaldehyde was added slowly with stirring. After complete addition, the reaction was heated on a steam-bath for 15 minutes, cooled, saturated with potassium carbonate, and the upper layer separated with the aid of ether. The ether layer was dried over potassium carbonate and distilled, yielding 60 g. (64%) of the methylolamine at 81° (22 mm.).
On treating with picric acid in ethanol, dimethylamine picrate formed, m.p. 159-160°C. The methylolamine, treated with 2,4-dinitrophenylhydrazine and hydrochloric acid in ethanol, gave a 2,4-dinitrophenylhydrazone, m.p. 92-93°, no depression of m.p. when mixed with a sample prepared from 3,5,5-trimethylhexaldehyde.
Benzaldehyde
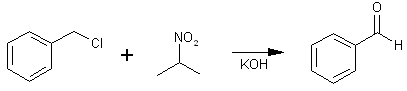
In a 200-ml. two-necked flask fitted with a mechanical stirrer and a reflux condenser, 14 g. (0.25 mole) of potassium hydroxide was dissolved in 25 ml. of water. 2-Nitropropane (22.2 g., 0.25 mole) was added, and the hot mixture stirred to a clear yellow solution. Benzyl chloride (31.7 g., 0.25 mole) was added, and the mixture stirred and heated under reflux for two hours. After cooling, the mixture was filtered through a glass wool plug to remove precipitated potassium bromide, washing with 50 ml. of ether. The ether layer was separated, washed with 25 ml. of water, dried over magnesium sulfate, and distilled. A small amount of acetoxime crystallized in the condenser (b.p. 52°C (18 mm.)), m.p. and mixed m.p. 61-62°C. The benzaldehyde was collected at 62°C (1 mm.), 13 g. (49%); phenylhydrazone, m.p. 157-158°C.7
2-Methyl-2-nitro-1-(para-nitrophenyl)-propane
Sodium hydroxide (12 g., 0.3 mole) was dissolved in 100 ml. of water and 26.7 g. (0.3 mole) of 2-nitropropane added and stirred to complete solution. To the solution was added 51.5 g. (0.3 mole) of para-nitrobenzyl chloride. The suspension was stirred and heated under reflux for 2.5 hours. After cooling, the supernatant water was decanted, the heavy oil taken up in 100 ml. of ether, washed twice with water, and the ether evaporated. The residual oil was crystallized from 100 ml. of hot ethanol, yielding 37.5 g. (56%) of crude product, m.p. 58-62°C. Recrystallization from ethanol raised the m.p. to 64-65°, no depression on mixing with an authentic specimen9.
Undecanal
A solution of 2.80 g. of potassium hydroxide and 4.55 g. of 2-nitropropane in 75 ml. of 95% ethanol was added dropwise in the course of 45 minutes to a refluxing solution of 11.75 g. of undecyl bromide (all reactants 0.05 M) in 50 ml. of 95% ethanol. Refluxing was continued for an additional 15 minutes. The solution was cooled and decanted from a deposit of sodium bromide; ethanol was removed in vacua, the residue taken up in 75 ml. of ether, washed with water (2x30 ml.) and dried over magnesium sulfate. After removal of ether, undecanal10a was distilled at 64-66°C/0.7mmHg, yield 7.3 g. (85%), n25 1.4500; 2,4-dinitrophenylhydrazone, mp 105-106°C; oxime, mp 71-72°C. A second preparation was started at room temperature and then heated under reflux for two hours, resulting in a similar yield.
This latter experiment illustrates a general procedure for monocarbonyl compounds. In the same manner there was prepared: dodecanal, bp 126-138°C/15mmHg10b;46% yield from dodecyl bromide; semicarbazone, mp 100-101°C10c; 2,4-dinitrophenylhydrazone, mp 102-103°C; 3,4-methylenedioxyphenylacetone, bp 110-111°C/0.8mmHg10d; 90% yield from 1-piperonyl-2-bromoethane (2-bromosafrole)11; semicarbazone, mp 158-159°C10d; 1,2-cyclohexadione, bp 80-81°C/16mmHg11 30% yield from 2-chlorocyclohexanone13 (reaction begun in ice-bath); bis-phenylhydrazone, mp 150-151°C.10f