Preparative column chromatography is one of the most valuable techniques for separating compounds in the synthetic laboratory. Unfortunately, this technique finds little use in the teaching laboratory because teachers must choose between an obsolete version, gravity column chromatography, and newer techniques such as flash chromatography and medium-pressure chromatography, which rely on expensive specialized glassware and require the use of hazardous pressurized columns. Another, little-known preparative technique, dry-column flash chromatography1, offers the separation power of the newer techniques but is considerably safer and less expensive. It is also easy to learn and use. Since dry-column flash chromatography is not covered in any of the popular undergraduate laboratory texts, we describe here an inexpensive adaptation of this technique suitable for the teaching laboratory, which we have used in our large general chemistry and organic chemistry classes.
General Procedure
The dry-column flash procedure resembles many other types of column chromatography in that one “packs” a column, loads the sample, and elutes the column. It is unique, however, in that the column (i) consists of a “dry” bed of silica gel placed in a sintered glass funnel, (ii) is eluted by using suction, and (iii) is drained dry after each fraction. These features make it much easier to pack the column, and students do not have to worry about their columns going “dry”. As a result, first-time users generally have no difficulty obtaining satisfactory results, and it is easy to get separations that rival those of analytical TLC.
Equipment
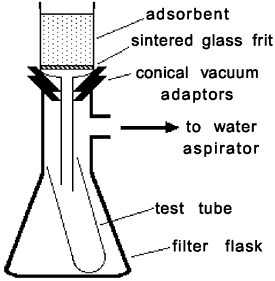
Figure 1.
Apparatus for dry-column flash chromatography
The column consists of silica gel (Merck grade 60, inhalation hazard!) packed inside a 4-cm diameter medium-porosity sintered glass funnel (Kimax 60 mL X 40 M). The rest of the apparatus consists of 16x150 mm test tubes and standard vacuum filtration glassware (Fig. 1). The eluting solvent needs to be a mixture of nonpolar and polar solvents; we use hexane and ethyl acetate for all experiments. The advantages of this combination are twofold: the two solvents have similar vapor pressures, and use of the same solvent pair in all experiments facilitates recycling.
Packing the Column
The funnel is loosely filled to the lip with dry silica gel (25-30g). The funnel is tapped gently to settle the powder and remove voids; then suction is applied (Fig. 1). Final packing involves pressing down on the silica carefully, but firmly, with a flat stopper or rubber cork while simultaneously applying full suction. One should end up with a reasonably level, compacted bed with about 1 cm of head space for the addition of the mixture and solvent fractions (if necessary, remove silica gel to create the necessary head space). The surface of the column need not be perfectly smooth.
Testing the Column
The column should be tested for voids and channels by carefully pouring hexane onto the bed (see following section, Protecting the Column) while simultaneously applying suction. The surface of the bed is kept covered with hexane until the solvent begins to flow out of the bottom of the funnel; then hexane addition is discontinued, and the column is sucked dry. If the packing has been done correctly the hexane front will descend the column in a horizontal line, and the final “dried” bed will have a uniform consistency. If the hexane advances significantly faster on one side, or if the bed develops channels or voids, the column should be sucked “bone” dry, repacked, and tested again.
Protecting the Column
Formation of a small crater in the middle of the column during solvent addition is unavoidable. The column can be protected, however, by covering it with a piece of filter paper and directing solvent onto this paper.
Loading the Sample Mixture
The simplest way to apply a sample mixture to the column is to dissolve the mixture in a small volume of hexane (e.g., 400 mg of sample dissolved in 2-3 mL of hexane), pour the solution evenly onto the column, and then apply suction. Hexane-insoluble sample mixtures can sometimes be applied in the form of a hexane-ethyl acetate mixture; the sample is mixed with 3–4 volumes of hexane, ethyl acetate is added dropwise until the sample just dissolves, and the resulting solution is added to the column in the normal way (it is wise to rinse the column with two or three portions of hexane before beginning the elution procedure). Yet another way to load hexane-insoluble samples is to pre-adsorb them onto a large excess of fresh silica gel. This is accomplished by dissolving the sample in a polar solvent, adding 5 mass equivalents of silica gel to create a slurry, and evaporating the solvent. The dry gel is then spread evenly on top of the column.
Eluting the Column
Once the sample has been loaded, the column is eluted using a series of hexane-ethyl acetate mixtures of increasing polarity. We have our students collect their solvent fractions directly in 16x150-mm test tubes placed inside the filtration flask (Fig. 1), so they do not have to rinse the flask (saving solvent), and do not run the risk of contaminating their samples. Elution is performed by applying full suction to the column and pouring ~25 mL of eluting solvent evenly onto the column (see Protecting the Column). Once the column is more or less dry, or the test tube is sufficiently full, the suction is released and the test tube is replaced. Some solvent is always lost through evaporation or adsorption, so it is not wise to make the solvent volume too small.
The polarity of the eluting solvent should be increased slowly but steadily from one fraction to the next. We use pure hexane as the initial solvent, and then steadily increase the amount of ethyl acetate in succeeding fractions by 1-2% increments until the mixture has completely eluted (larger increases are possible with easier mixtures, such as ferrocene-acetylferrocene). Mixing the solvent fractions turns out to be the most time-consuming part of the procedure, but the time required can be significantly reduced by following this four-step procedure:
Step 1: 100 mL of the solvent mixture needed for fraction X is prepared in a 100-mL graduated cylinder, and 25 mL of this mixture is used for fraction X;
Step 2: A 'squirt' of ethyl acetate (0.5-1 mL) is added to the remaining solvent via disposable pipet, and 25 mL of this mixture is used for fraction X+1;
Step 3-4: Step #2 is repeated two more times to give fractions X+2 and X+3.
At this point, one returns to Step 1 using a new, more polar solvent mixture (copies of our exact 'solvent schedule', starting with pure hexane (Fraction #1) and ending with 60:40 hexane:ethyl acetate (Fraction #24), are available upon request).
Sample Detection
If the sample components are colored (e.g., ferrocene-acetylferrocene), the progress of the separation can be monitored visually. A more general procedure, however, is to collect 12-16 fractions and then analyze them simultaneously by TLC.
Applications
We have successfully used this technique to separate mixtures ranging from 150 mg to 1 g. The separations can be of near-TLC quality, even in relatively inexperienced hands, provided the solvent polarity is increased slowly. The separations our students perform include: ferrocene-acetylferrocene2, cis- and trans-3,3,5-trimethylcyclohexanol (prepared by NaBH4 reduction of the ketone), and the separation of a colorless unknown binary mixture (one component of which is either propiophenone, ethyl benzoate, or anethole, and the other component either ortho-diethyl phthalate, ortho-dimethyl phthalate, or cinnamyl alcohol). Complete resolution is normally achieved in each of these cases; and separation of the diastereomeric cyclohexanols is especially noteworthy because the NaBH4 reduction is moderately diastereoselective, and students can easily distinguish between the two resolved stereoisomers by NMR (details of all of the experiments can be obtained from the authors upon request). Although the original description of the technique warns of possible oxidative decomposition1, we have never observed this, even in the case of anethole and cinnamyl alcohol.