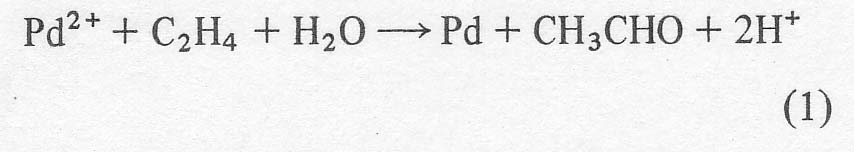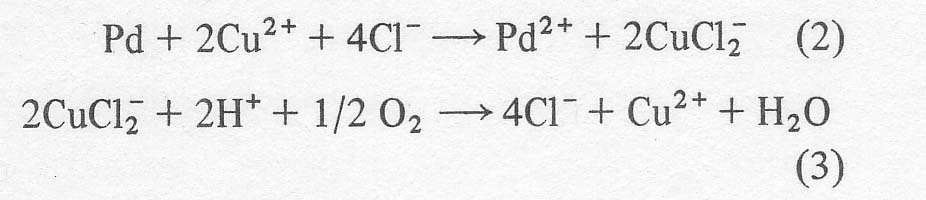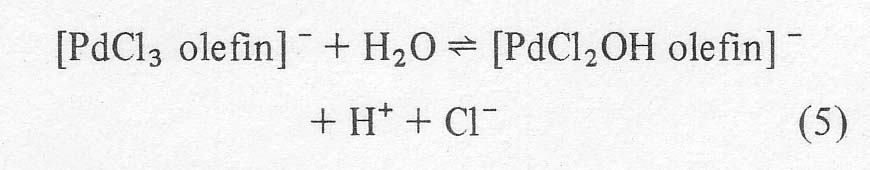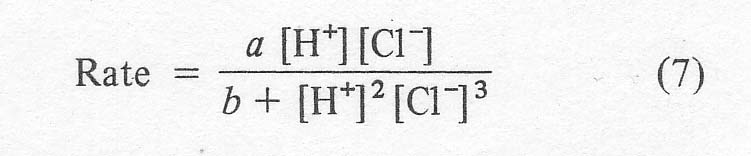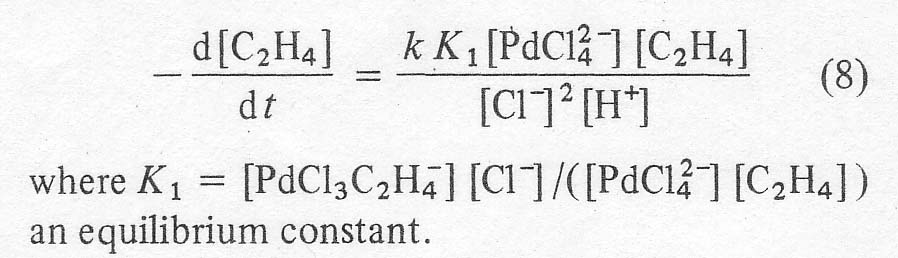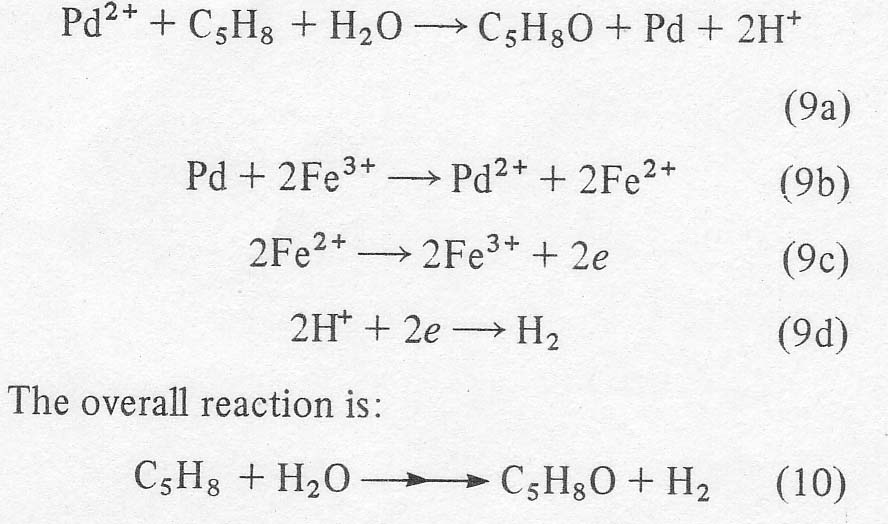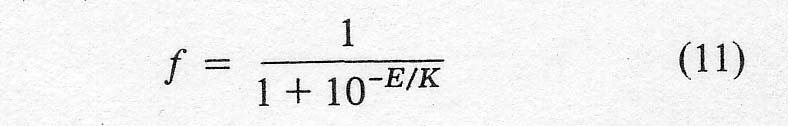Electrochemical process for converting olefins to ketonesby H.H. Horowitz, Journal of Applied Electrochemistry 14, 779-790 (1984)HTML by ScoobyDoo [ Back to the Chemistry Archive ] Olefins higher than 3-C were converted to ketones using aqueous chloropalladate ion as oxidant. The chloride ion content of the solution was kept at very low levels relative to normal usage to increase significantly the reaction rates of these higher olefins. The reduced palladium was reoxidized electrochemically using the ferric/ferrous couple as the transfer agent. Using cyclopentene as a model compound the ionic strength, the acidity, the concentration of palladium, the means of extracting product and the means of adding olefin were all adjusted as to achieve about 70% current selectivity with minimal palladium inventory. There were indications of even better behaviour with several open chain ketones.
1.
Introduction This
paper discusses a means of converting olefins higher in molecular weight than
ethylene into the corresponding ketones via an electrochemical version of the
Wacker process. In the latter process [ 1-3 ], the olefin, ethylene or
sometimes propylene, is bubbled through an aqueous solution of palladium
chloride where it is converted to the corresponding carbonyl compound,
acetaldehyde or acetone in these cases. With no oxidant present, palladium
metal would eventually precipitate out.
However,
cupric chloride and hydrochloric acid are also present in the solution to
reoxidize the reduced palladium and form the dichlorocuprate (1) anion. An
excess of hydrochloric acid is required to render the cuprous complex thermodynamically
stable. In the absence of excess chloride, cuprous ion would dismutate to
copper metal and cupric ion. Finally, the cupric ion is regenerated by blowing
air or oxygen through the mixture so that the net result is that olefin plus
oxygen yield carbonyl, and all other reagents are recycled.
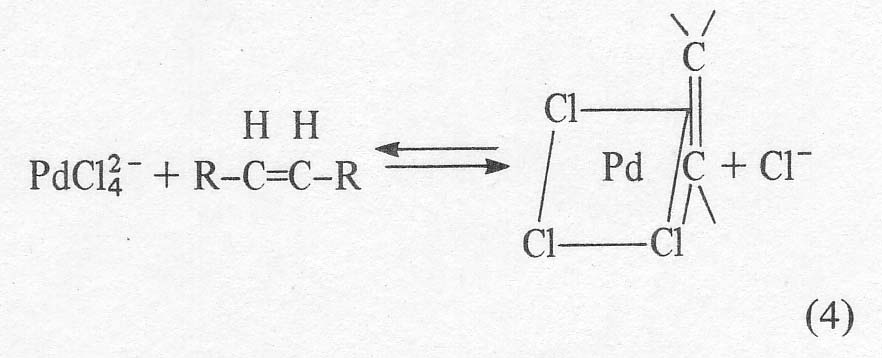
Kinetic
and mechanistic work on the process [4-7] reveals that the chemical Wacker
process runs despite the fact that the chloride required to stabilize the
cuprous ion is a strong inhibitor for the reaction. There are at least two
steps in the mechanism where it has been postulated
Actually,
this soluble ion is postulated by analogy with the platinum analogue, Zeise's
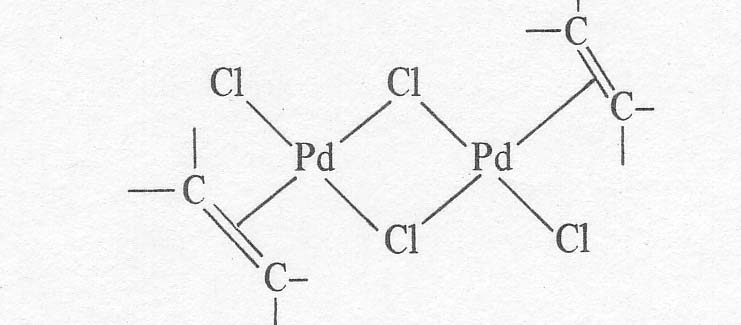
salt, KPtCl3C2H4. The solid palladium compounds that have actually been
isolated are neutral dimers of the apparent structure given below [8].
Kinetic
and isotopic data suggest that after the complex forms, the next step involves
reaction with water to produce an aqua complex which ionizes to a hydroxy
complex releasing more chloride and hydrogen ions:
In the absence of excess chloride, the same complex may be arrived at by the
reaction of an aqua complex of PdCl2OH(H20)- with an olefin molecule. At
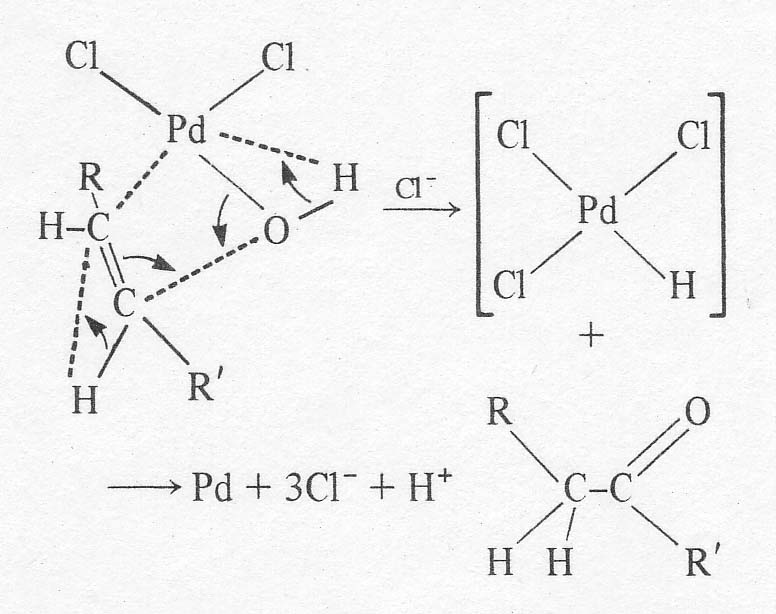
this point, a relatively slow rearrangement of the complex occurs whose
details are in doubt. A good discussion of the mechanism is given by Aguilo
[9]. There may be a shift of the OH group from a trans- to a cis-position
relative to the olefin. Then, the complexed olefin is apparently attacked at
one end of the double bond by the neighbouring OH group causing the organic
ligand to shift from p-bonding
to s-bonding to the
palladium. A hydride is shifted from the attacked carbon to its neighbour, and
a molecule of the carbonyl compound splits off the complex. The palladium is
left in a reduced form, possibly as a hydride (postulated by Halpern [10])
which soon decomposes to the metal:
The most certain step in the above scheme is the shift of the hydrogen. Radiotracer studies have confirmed that the hydrogens on the carbon a to the carbonyl come from the olefin and not from the solvent. The hydride shift is not involved in the rate determining step, however, based on the lack of a kinetic isotope effect with C2D4. The reaction shows 'carbonium ion character' in that the Markonikov product is obtained. That is, ketones are formed from a-olefins in great preference to aldehydes. It is possible that the Pd-C sbond is partially ionic, leaving a slightly positively charged carbon. The effect of hydrocarbon structure on reaction rate is small, so that a free carbonium ion is probably not involved in the rate determining step. In any case, the important mechanistic point for the purposes of this work is that the reaction is sharply inhibited by chloride ion. The original workers [4] give an empirical rate expression for the ethylene reaction as:
where
the bracketed terms appear to signify the total solution contents. This
expression shows the initial rise of rate at low chloride contents and the
subsequent decline in rate with the square of the chloride content at higher
concentrations. A similar relationship was shown for n-butene in 1972 [11].
Another rate study by Henry [5] gives:
The
bracketed terms indicate the concentrations of the actual species. This
expression is not suitable for systems containing less than 3-4 moles of total
chloride per palladium, but again shows the sharp inhibiting effect of
chloride at higher concentrations. Apparently,
ethylene is so reactive that it can withstand this impediment; however,
reactivity drops with olefin size as given by the data of Henry at 25� C
(Table 1). This is one of the
A
second inherent drawback associated with the use of air oxidation in acidic
copper halide solutions is the formation of chlorinated by-products. Except
for its palladium content the solution used for air regeneration in the Wacker
process has the same ingredients as those used in the Kellogg olefin-oxyhydrochlorination
process [12]. The by-products are apparently troublesome to remove, as
indicated by the issue of various patents concerned with their separation
[13]. In
this paper, we consider an electrochemical process that permits the reaction
to be run in the absence of excess chloride and oxygen. The reduced palladium,
in whatever form it exists before it precipitates (shown above as PdCl3H2-),
is reacted with an oxidant not requiring chloride for stability, such as
ferric ion. The resulting ferrous ion is reoxidized electrochemically at an
anode inserted in the aqueous phase. At the cathode conditions are arranged so
that hydrogen is liberated. The complete scheme is: All
other reagents recycle. This idea was actually suggested in 1967 by
researchers at Hoechst [14]. The patent was apparently never implemented
commercially, perhaps because it advocated total elimination of chloride ion,
and might, therefore, have been inoperable. The
aim of this experimental programme was to determine whether such a scheme
was feasible: whether the product could be isolated in high yield, whether
the palladium inventory could be kept economically low and whether the palladium
could be successfully recycled, and so on. The olefin chosen for the model
study was cyclopentene, though others were subsequently examined. 2.
Experimental details The
investigation was carried out in a simple glass apparatus, which was gradually
modified and enlarged with other glass ancillaries as the variables were
analysed and the process took shape. The basic cells consisted of a jacketed
anode compartment connected via a glass frit to a side arm containing the
cathode. The cathode was a platinum wire used for hydrogen evolution. The
anode compartments were of 250 or 500 cm� capacity. A 5 cm x 5 cm gold-plated
tantalum screen served as the anode for most of the experiments, although a 5
cm� platinum screen was used in preliminary runs. The cell was also equipped
with a gold wire probe and a reference electrode containing a gold wire,
immersed in an equimolar mixture of ferrous and ferric perchlorates in 0.3 M
perchloric acid. This was used to monitor the potential of the ferrous-ferric
couple during measurements of the rate of reaction or during regenerations.
The
electrolyte used was 0.3 N perchloric acid (9a) containing varying
concentrations of ferric and ferrous perchlorate. The latter were either
purchased or prepared by dissolving high purity iron in more concentrated
perchloric acid to form the ferrous salt and treating with dilute hydrogen
peroxide to form the ferric salt. It was subsequently found that during this
iron dissolution step some reduction of perchlorate to chloride occurred to
the extent of 0.05-0.15 moles of Cl per mole of Fe2+. Since the concentration
of iron ions in solution eventually preferred was 0.2 M, an added chloride ion
concentration of approximately 0.02 M was always present. Fortunately, this
concentration kept the reaction rates close to the maximum value. In
later experiments, the glass apparatus was expanded to permit solvent
extraction of the cyclopentanone product. Solvents were employed in a unit
comprising a boiler, which sent the extractant vapours through a fractionating
column to a condenser, from which the liquid was allowed to flow down to a dip
leg to the bottom of the electrolyte chamber. Solvent collected above the
electrolyte and overflowed through a side arm to the middle of the
fractionating column, and was recycled. The
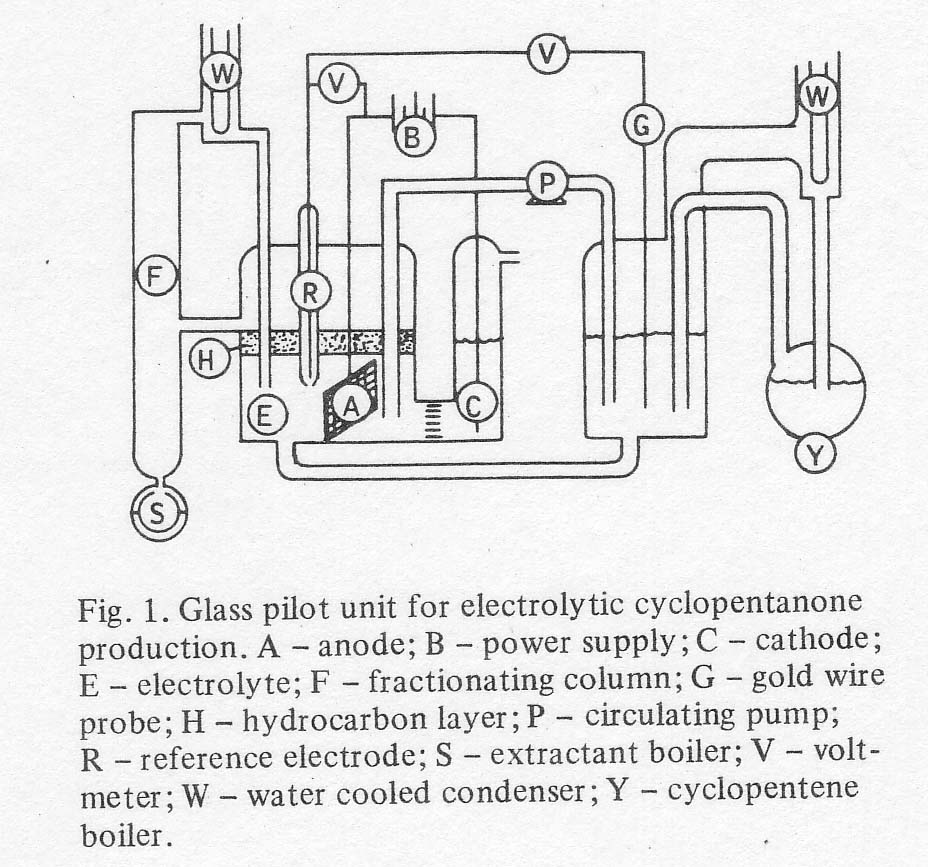
total 'pilot unit' now contained two main vessels, a reactor and a
regeneratorie extractor, as shown in Fig. 1. The continuous extractor
described above is shown on the left in the figure, using 2-methylpentane as
the extractant. The extractant passed through the left hand vessel containing
electrolyte (HClO4, Fe 2+ and Fe 3+ perchlorates and PdCl2). The
electrochemical regeneration of ferric ion was also carried out in this vessel
by means of a gold-plated tantalum screen anode and a platinum wire cathode in
a chamber isolated by a coarse glass frit. Gaseous cyclopentane was fed from a
second boiler with a liquid return line, into the reactor vessel. Electrolyte
was circulated between the two vessels by means of a Teflon pump and return
line of glass with polypropylene fittings. Power was supplied by a D.C.
galvanostat. The voltages in both cells were read vs a 1/1 ferric/ferrous
electrode. The circulation rate was maintained rapid enough so that the
difference in voltage between the two reactors corresponded to a difference in
ferric ion concentration of no more than 2% of the total iron concentration.
Approximately 40 runs were made from which the data shown in the following
section was taken. During
measurement of the rates of the reaction of the olefin with palladous ion, no
electrochemical regeneration was carried out; the effect of the reaction was
simply to reduce the ferric ion to the ferrous ion. The fraction of the total
iron present as ferric, f, at any time was calculated from the voltage, E,
between the gold wire and the reference electrode using a modified form of the
Nernst equation,
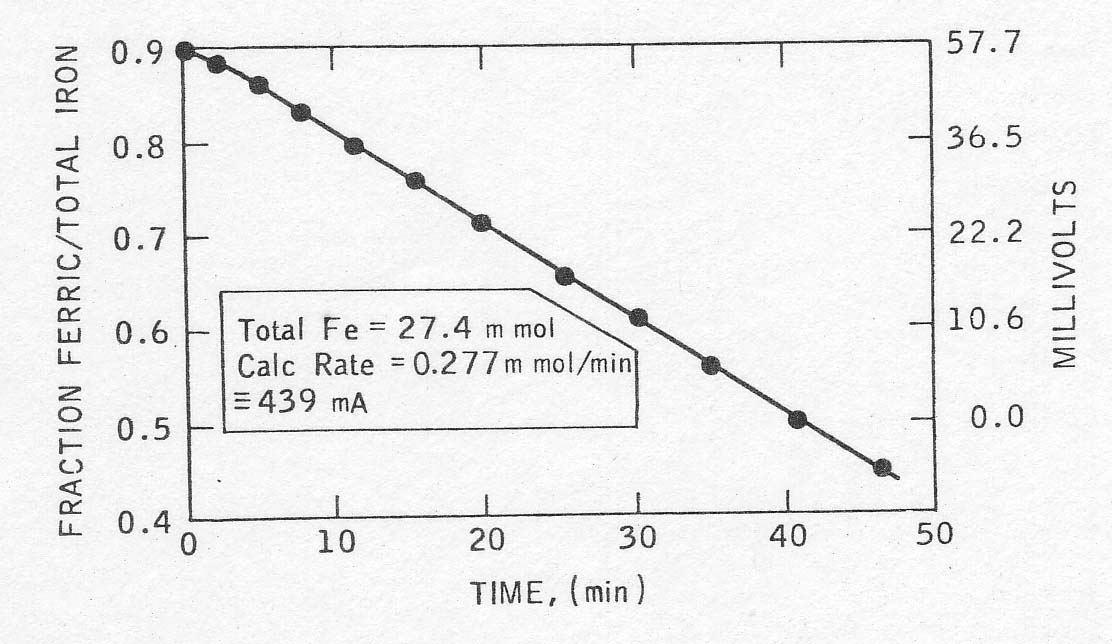
A
good example of a rate plot thus determined is given in Fig. 2. From the slope
of the plot and the total iron concentration, the rate of the reaction was
calculated. It could be expressed in terms of current by multiplication by
Faraday's number, and further normalized to current per gram of palladium for
comparison with other runs. In one case, the rate calculated from the open
circuit potential drop was verified approximately by determining the
regeneration current necessary to hold the solution potential constant (see
Table 2).
Routine
analysis for cyclopentanone was carried out by extracting the electrolyte
with ethyl ether, evaporating some of the ether through a fractionating column
and examining the residue in a gas-liquid chromatograph using one or more
internal standards. The gas chrornatographs showed a clean base line, indicating
greater than 99% purity for the product in the extract. In one case, the product
was isolated as the 2,4dinitrophenylhydrazone and proved to have the same
melting point and mixed melting point as a sample of authentic cyclopentanone
2,4dinitrophenylhydrazone. 3.
Experimental results and discussion
3.1
Acid concentration The
acid concentration for the majority of the study was chosen on the basis of
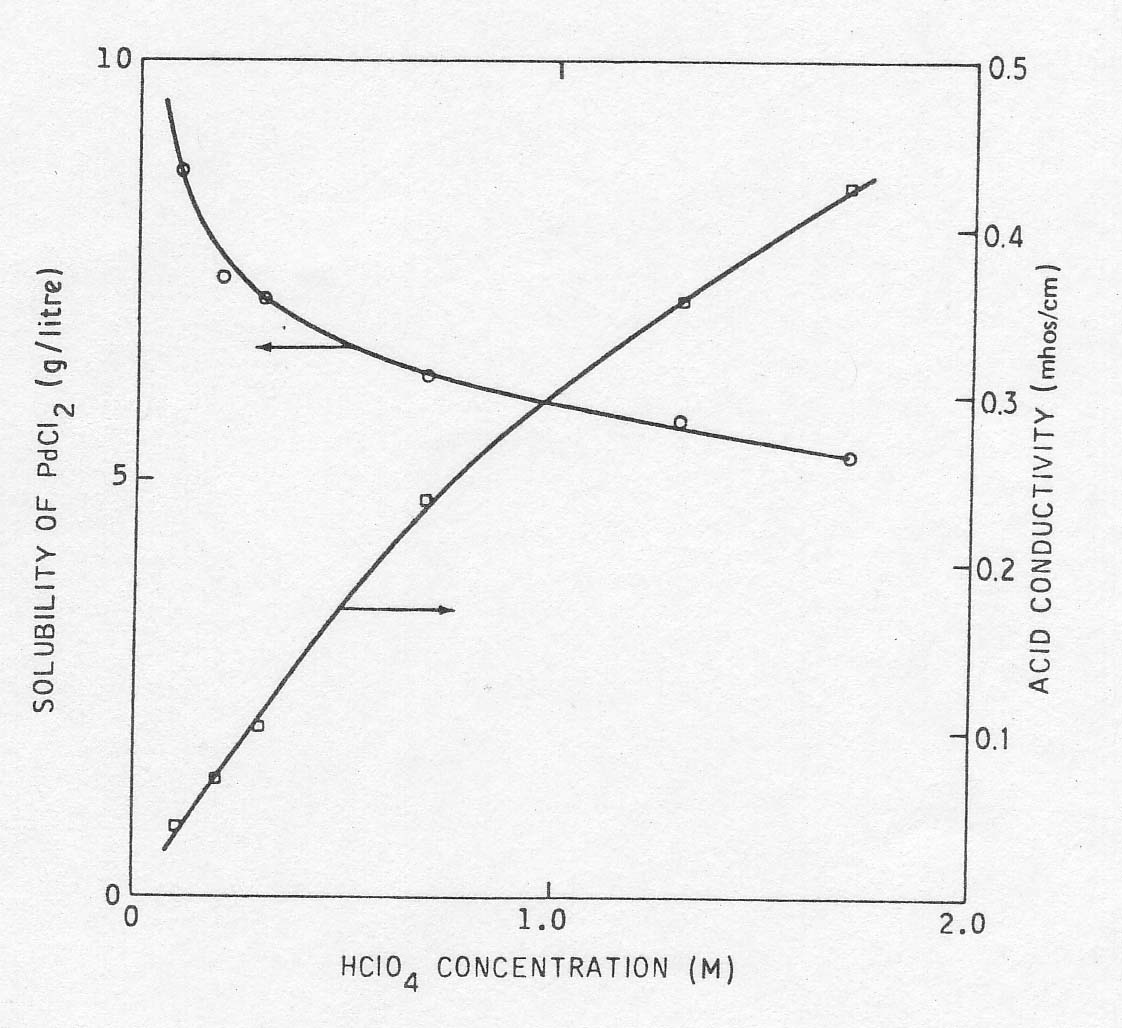
its effect on the solubility of PdCl2 and on the electrical conductivity. The
experimental results are shown in Fig. 3. It will be seen that the solubility
of PdCl2 decreases with acid concentration. To permit the maximum variance in
palladium concentration while maintaining reasonable electrolyte conductivity,
a concentration of 0.3 M HCl04 was chosen. However, because of the high
reaction rates ultimately obtained, lower palladium concentrations could have
been tolerated and higher acidity would have been desirable. This would have
improved conductivity, reduced concentration polarization in the cathode
chamber and, according to Equation 7, would have accelerated the reaction at
low chloride concentrations.
Fig
3 The effect of HClO4 concentration on conductivity and PdCl2 solubility. In
a few preliminary experiments performed at 350 C with liquid cyclopentene with
various electrolytes, it was verified that the perchlorate electrolyte did
indeed provide a large rate advantage over electrolytes containing substantial
amounts of chloride as shown in Table 3. It
will be seen that the rates are of the order of sixty times as great in the
perchlorate solution as in the others and that the difference is not due to
the presence of perchlorate, but the absence of chloride.
3.3
Effects of ionic strength and chloride content Early
attempts to use the ferric/ferrous redox couple at initial ferric concentrations
of 0.6 M led to precipitate formation in the solution after the cyclopentene
was introduced. This precipitation slowly generated palladium black, if wet
with dilute perchloric acid or dissolved in aqueous acetone. Based on literature
reports of the isolation of such complexes, it was probably the (PdC12 olefin)�
dimer referred to in the Introduction. It is interesting that this complex
could be dissolved in 1.3 N hydrochloric acid, where it yielded no palladium
precipitate in 24 h, again attesting to the inhibitory properties of chloride
ion. The material was not isolated in a pure enough form to be analysed, however. Reducing
the ferric ion concentrati6n prevented the precipitation of the complex.
Apparently, the palladium-cyclopentene complex, with its high hydrocarbon
content, and therefore borderline solubility in water, was salted out by the
high ionic strength provided by the ferric ion. (A solution of 0.6 M in ferric
perchlorate contributes 1/2 (0.6 x 9) + 3 x 0.6 = 3.6 mol 1-� to the ionic
strength.) Reducing the iron concentration and thereby the ionic strength,
avoided precipitate formation and also increased the rate of the reaction
somewhat (Table 4).
The
effects of additions of chloride ion to the 3.4
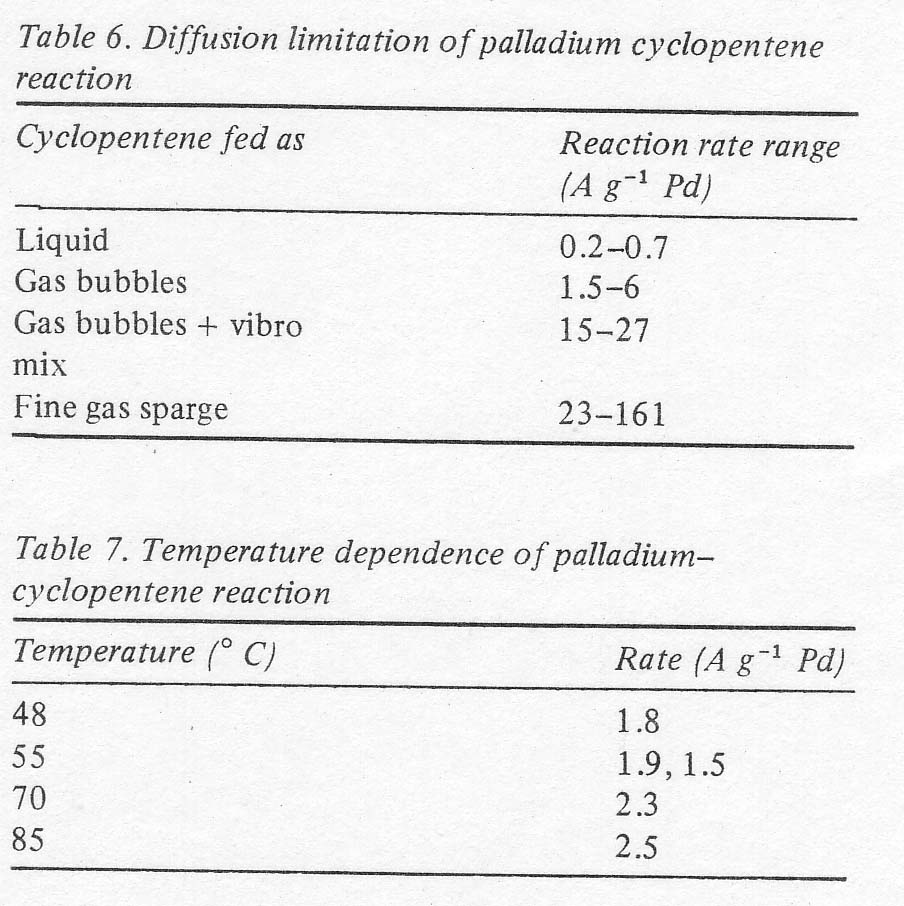
Diffusion limitation Under
conditions of reduced ferric and chloride contents, the factor having the
largest effect on the reaction rate was the mode of feeding the olefin. Admitting
it as gas bubbles gave a higher rate than simply stirring the liquid cyclopentene.
Adding a mechanical mixer along with the gas feed increased the rate further,
and finally sparging the olefin as a fine foam gave the highest rates obtained;
in the order of 100 A g -� (Table 6) as contrasted to the values of 600 mA
g-� in Table 5. The practical significance of this is that the palladium inventory
required for commercial production becomes minimal at the highest rates. For
example, at 100 A g -� an inventory of
5700 g Pd is adequate for a production rate of ten million pounds of
cyclopentanone per year at 70% selectivity. The investment cost of palladium
should, therefore, amount to well under one cent per pound of ketone produced.
The range of values shown in Table 6 is partially due to other variables studied,
namely temperature and concentration, and partly due to the experimental variability
connected with a diffusion limited reaction. These data indicate that the
reaction itself is very fast and that the rate is limited only by the diffusion
of the olefin into the liquid phase or by diffusion of the olefin complex
from the edge of the bubbles into the bulk. The temperature dependence under
these conditions was very low, and the reproducibility was not good (� 10%)
again as might be expected for a diffusion limitation (Table 7).
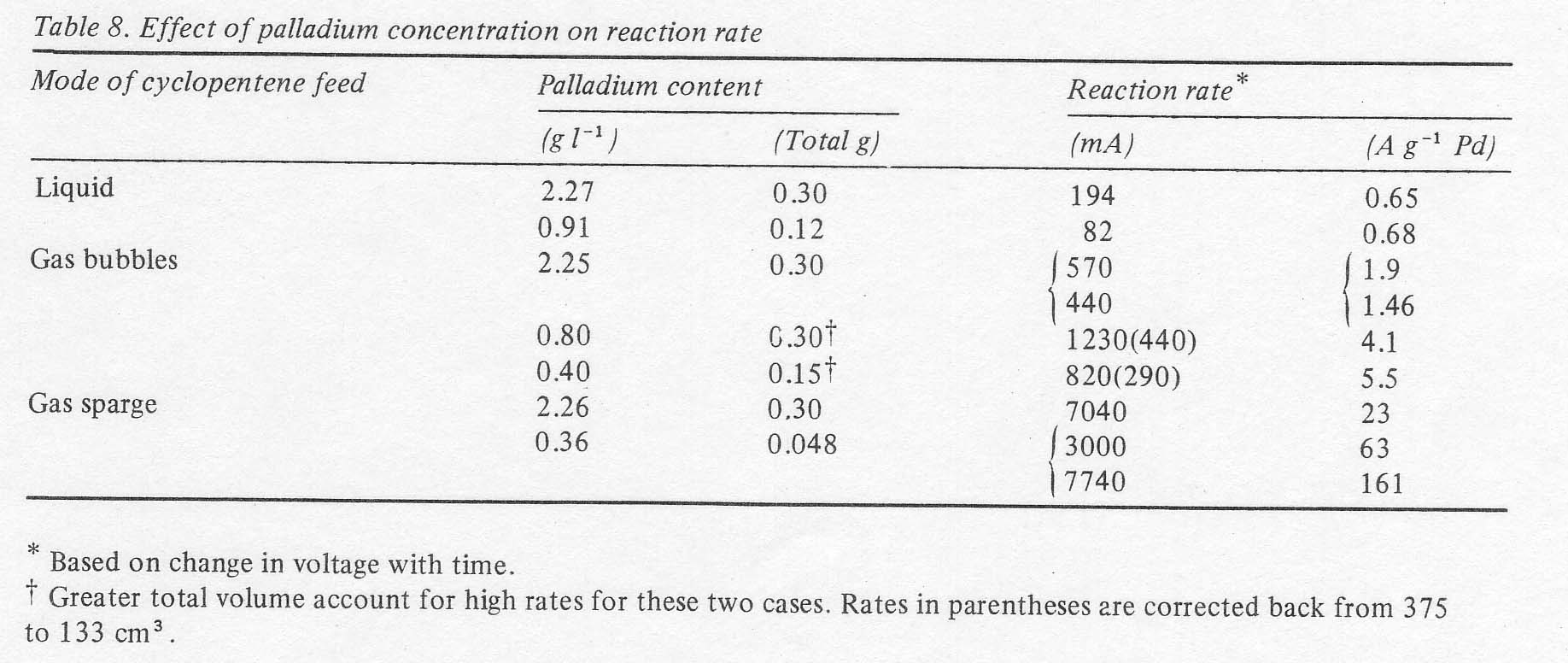
It
is interesting that changes in palladous ion concentration produced nearly
proportionate changes in reaction rate, thus giving little change in activity
per gram of palladium, at conditions of poor mass transport. Where olefin
contact is better and the rates are higher, it is advantageous to use lower
concentrations of palladium dichloride for the reaction rate tends to reach a
limit at high palladium concentrations (Table 8). In
any case, the rates are high enough that the palladium inventory is no
problem. 3.5
Product yield and stability The yields of ketone varied from 25 to 74%, based on the total current passed and/or oxidant reduced, due to slow further reactions of the cyclopentanone in the electrolyte. Where the rates were low and the runs took 4-5 h to complete, current selectivities were only about 30%.
But
where continuous extraction was carried out or the rate was high and runs
short, current selectivities of 70% were observed (Table 9). The
rates of oxidation of the ketone were measured in separate experiments simply
by adding it to the electrolyte containing the palladium chloride, perchloric
acid and ferric perchlorate, and measuring the rate of ferric reduction from
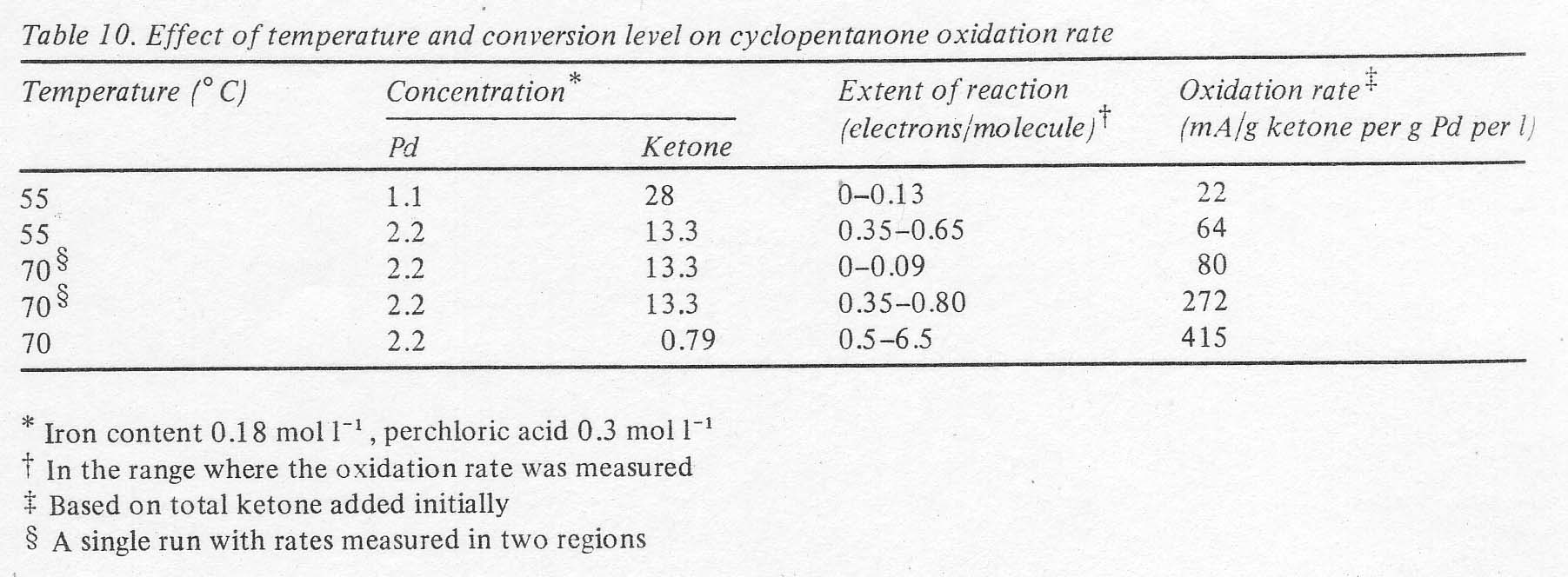
the open circuit voltage decline. The rates were not constant in a single run,
but appeared to increase with time, as if the initially formed products were
more reactive than the starting ketone. At a given conversion level, the rates
were fairly sensitive to temperature, as shown in Table 10. Not
all the oxidation was caused by the palladium. At 70� C, the oxidation rate
due to ferric alone in the absence of palladium was one fourth that of the
oxidation rate of the total system. In the absence of ferric ion palladous ion
oxidized cyclopentanone readily, as evidenced by a rapid drop in potential and
precipitation of palladium metal. No rate data were taken under the latter
conditions, however. The
ketone oxidation reaction probably does not go to a single intermediate, but
appears to proceed largely to C02. In a long term run with an excess of
oxidants, the reaction rate declined with time until eventually 20 equivs of
oxidant were consumed per mole of ketone. Complete conversion to C02 would
require 26 equivs per mole. In a subsequent run, considerable C02 was trapped
in an Ascarite tube, though the results were not quantitative. This result is
favourable, for it means that the current used in oxidizing ketone corresponds
to very little wasted ketone. A current selectivity of 70% corresponds to a
product selecitivity of 98%, assuming the side oxidation proceeds all the way
to C02. Due
to the complexity of the ketone side reactions, the above data may be used
only as a rough index of the rate of product loss via side reactions. Using
the maximum rate of ketone oxidation at 55� C, and the observed rate of
olefin oxidation, one calculates current selectivities to product which are
higher than those actually observed. Thus, the yields must also be lowered by
acid catalysed condensation reactions and " physical losses, not detected
by this method. In any case, the need for continuous extraction to reduce the
concentration of ketone in the electrolyte at any time, is clearly indicated. The
susceptibility to further oxidation is unusually severe for cyclopentanone and
not typical of open chain ketones as will be discussed later. 3.6.
Continuous extraction To operate for long periods of time with product removal the continuous extraction set up described in Section 2 (Fig. 1) was used. The requirements for the extraction solvent proved to be quite stringent. Cyclopentene itself extracted the ketone from the aqueous phase quite efficiently and was at the same time the fed olefin. However, as has been discussed, rates of reaction were very low unless the olefin was fed as a gas, which made it impossible to use pure cyclopentene as extractant.
Furthermore,
the cyclopentene-palladium complex was soluble to a small extent in
cyclopentene, so that in long runs black palladium was visible in the solvent
boiler. It was possible to operate at 55� C with liquid cyclopentene (b.p. = 44� C)
with a mixed extractant containing cyclopentene plus a higher boiling liquid
such as benzene or cyclohexane. However, the organic layer tended to be rich
in cyclopentene and the palladium complex was again extracted. Finally, it was realized that the extractant would have to be a saturated
hydrocarbon and that the cyclopentene gas would have to be fed in a separate
vessel so that it would not accumulate in the extractant stream. Since the
reaction is diffusion limited, the amount of olefin dissolved in the
electrolyte and thereby able to reach the extractant would be negligible. Cyclohexane and 2-methylpentane were tried. Their ability to extract
cyclopentanone from aqueous acid into a hydrocarbon layer was inferior to that
of cyclopentene, but still tolerable (Table 11). The methylpentane was finally selected as best since its lower boiling point,
60� C (cf. 81� C for cyclohexane), reduced the rate of evaporation of the
product (b.p. = 131� C) from the extractant boiler. Its use completely
eliminated the palladium extraction problem. The rate of accumulation of
ketone in the boiler was measured by gas chromatography relative to internal
standards which had been placed in the boiler at the start of the experiments.
In trial extraction experiments, the contacting efficiency was calculated to
be about one quarter to one third of that if saturation were always complete.
This, coupled with the limited solvent recycle rates and the low distribution
constant, caused the extraction half-life to be estimated at about 2 h. Thus,
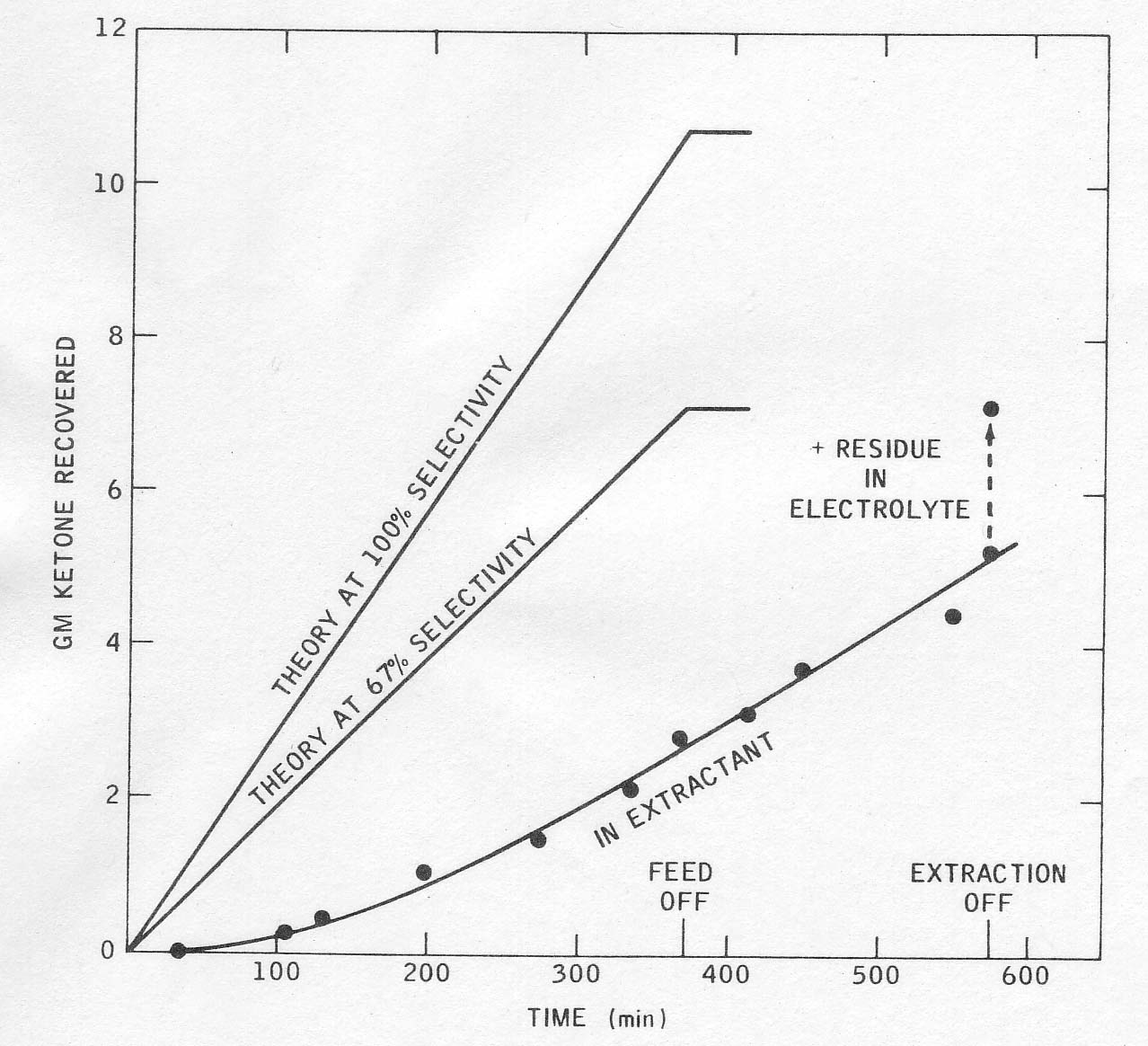
in continuous runs the product concentration in the electrolyte built up
slowly until the rate of extraction matched the rate of production of
cyclopentanone. Product accumulation in the solvent boiler lagged considerably
behind its production and continued after the reaction stopped. All of this
could be improved in a larger plant of pilot unit, and the yields thereby
increased. 3.7.
Cell voltage requirements
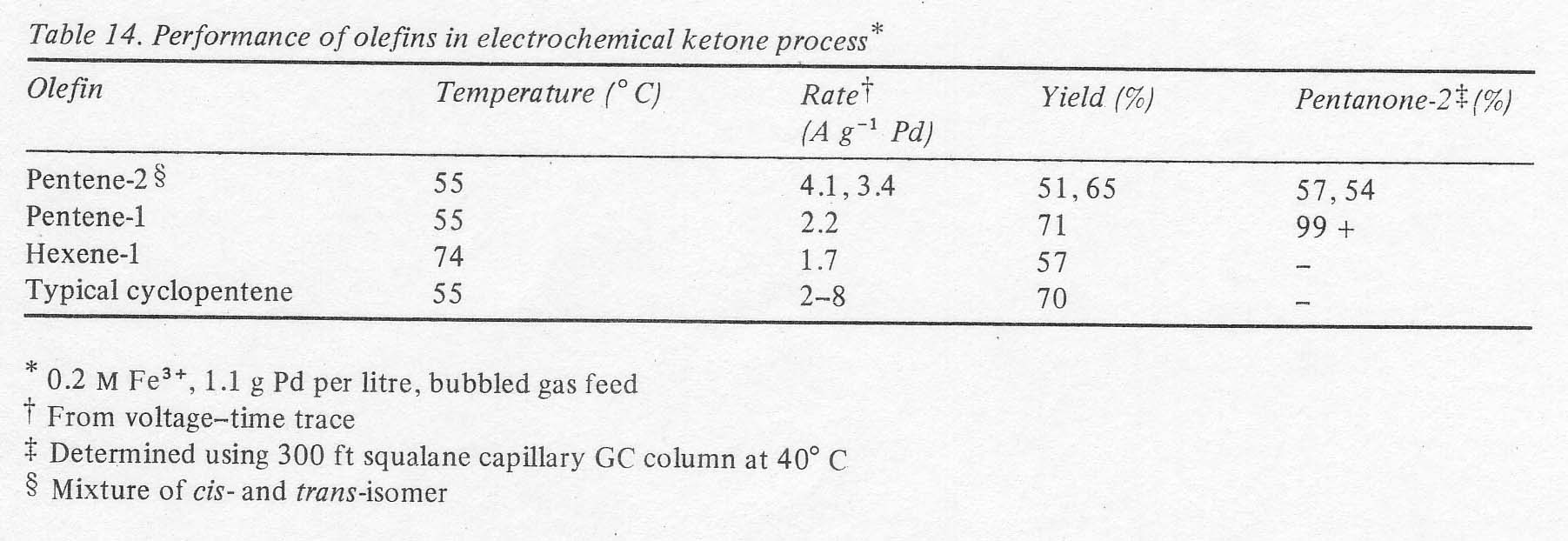
The possibility of manufacturing other ketones by this electrochemical process was examined briefly. It was first of all verified that open chain ketones are at least an order of magnitude more stable to oxidation than cyclopentanone in the acidic palladous-ferric solution (Table 13). The results suggest that yields should be higher and the necessity for continuous extraction is less for ketones other than cyclopentanone from the standpoint of oxidation stability alone. Of course, the possibility of acid catalysed condensations, not measured by the above procedure, still makes continuous extraction mandatory. The yields and reaction rates with open chain olefins were of the same order of magnitude as cyclopentene (Table 14). This was determined in batch experiments run without continuous liquid extraction; using only the two right-hand vessels in Fig. 1. Most of the product ketone was found in the olefin boiler due to the exceptionally high activity coefficients of C5 and C6 ketones in aqueous solutions. Thus, the product was being removed continuously. The isomer distributions shown in the last column verify that terminal olefins form essentially no aldehydes, that double bonds do not migrate in this process and that internal olefins form the two possible ketones in approximately the manner expected from the carbonium ion character of the reaction. In the case of pentene-2 a more sensitive chromatograph was used, and a number of minor peaks were found. Their total area amounted to less than 1% of the major product peaks, however.
The reaction was also carried out on the branched olefin, 2-methyl butene-2, a likely contaminant of cyclopentene. Gas-liquid chromatography on the resultant extract showed at least six different products in comparable amounts, whose identity was not further investigated. Smidt [3] also indicated that olefins bearing no hydrogen on one of the double-bonded carbons react slowly, undergoing hydration, other oxidations and rearrangements, and later, cleavage. lsobutylene, for example, yielded tertiary butanol along with acetone, isobutyraldehyde, a-methyl acrolein and methyl ethyl ketone. Branched olefins are best eliminated from the feed stream. 4. Conclusion
This electrochemical chloropalladate
process looks promising for small plants converting C4 and higher linear olefins
to the corresponding ketones, cases where the corresponding air oxidation
process has not been commercialised. The advantage of the electrochemical
process lies in its avoidance of excess chloride, which leads to slow rates
and chlorinated by-products. The process has been demonstrated in a continuous
laboratory unit with good current selectivities and maintenance of high catalyst
activity. The process cost remains to be investigated. It is known that the
palladium inventory can be very low, but the electrode cost and the materials
of construction have not been determined. The details of product recovery
via extraction or stripping would have to be worked out for each olefin one
desires to oxidize. Further studies would also have to be carried out to choose
the
optimum acid type and concentration based on considerations of safety and
total cell voltage drop. Presumably, dilute sulphuric acid could replace the
perchloric acid used in this study. Cells would have to be designed to
optimise the current densities for ferrous oxidation, to reduce the cathodic
reduction of ferric ion and to minimize the voltage drop. None of these
problems appear to present any great difficulties. References [1]
J.Smidt,W.Hafner,R.Jira,J.Sedlmeier,R. Sieber, R. Ruttinger and H.
Kojer, Angew. Chem. 71 176 (1959). [3]
J. Smidt, Chem. Ind. (1962) p. 54. [4]
R. Era, J. Sedlernier and J. Smidt, Ann. Chem. 693 (1966)99. [5]
P. M. Henry, J. Amer. Chem. Soc. 86 (1964) 3246. [6]
Idem, ibid. 88 (1966) 1595. [7]
M.N. Vargaftik, 1. 1. Moiseev and Y. K. Sirkin, Dokl. Akad. Nauk. SSSR
147 (1962) 399. [8]
G. F. Pregaglia, M. Donati and F. Conti, Chem.Ind, 46 (1966) 1923. [9]
A. Aguilo,Adv. Organomet. Chem. 5 (1967) 321. [10]
J. Halpern, Chem. Eng. News October 31 (1966) p. 74. [11]
H. Hasegawa and M. Iriuchijima, US Patent 3 701810, (1972). [12]
M. L. Spector, H. Heinemann and K. D. Miller, Ind. Eng. Chem. Process
Des. Dev. 6 (1967) 327. [13]
G. The Uig, E. Weber and H. Steinro etter, US Patent 3 330 741 (1967). [14]
0. Heuse, E. Weber and R. Wirtz, Canadian Patent 768 13 7 (1967). |