Synthesis of DOB without LAHSynthesis of DOB with NaBH4 and Al/Hgby Iudexk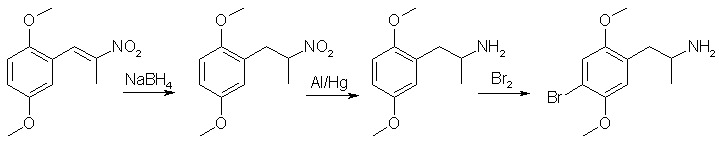 2.84g [75mmol] NaBH4 and 1g silica gel was added to 30mL anhydr IPA and cooled to 0°C in an ice/salt bath. A solution of 4.15g [18.6mmol] 2,5-dimethoxyphenyl-2- nitropropene in 25mL t-Bu-O-Me was then added dropwise with stirring at a rate such that rxn temp. did not exceed 5C. Stirring was then continued at room temp. for 6hrs. The rxn mixture was acidified by the slow addition of 50mL 32% HCl. 100mg HgCl2 was then added and dissolved. 3.011g [112mmol] aluminium foil was then added slowly. After all Al had disappeared, rxn mixture was filtered, tBu-O-Me/IPA evaporated, to give red aq. layer, which was washed w/DCM, some of the red coloring being removed, DCM extracts perfectly clear. This was then basified, extracted w/DCM, DCM washed w/H2O, dried over MgSO4 and evaporated to yield 1.205g 2,5-DMA as a pale yellow oil. This oil was dissolved in 7.6mL GAA, and under sitrring 1.135g Br2 in 2.5mL GAA was added dropwise over 4mins. The rxn mixture was left to stir for 3hrs, poured into 125mL H2O, washed w/tBu-O-Me, basified and extracted w/DCM. The DCM was evaporated [forgot to add boiling chip, flask overboiled, some of product lost] to give an amber oil, which was dissolved in 75mL Et2O and gassed w/HCl [CaCl2 H2SO4] under stirring. Flask was put in a freezer for 1hr, then filtered [filtrate pH = 5.5] and air-dried to yield 0.69g 2,5-dimethoxy-4-bromoamphetamine hydrochloride as a white crystalline powder. Notes
Bioassay of product, which I am currently bioassaying as I type this, is fucking unbelievable [nice] Synthesis of DOB with NaBH4 and Al/Hgby Dr Gonzo10g of 2,5-dimethoxy-beta-nitropropene was placed in a 300 ml beaker with magnetic stirbar and 250 ml methanol, and stirred for a few minutes to dissolve as much as possible. Sloooowly... 50 mg at a time... 2.2 g NaBH4 was dropped into the sirring solution, with moderate effervescence with every addition. Addition of all the NaBH4 took less than 30 minutes. After a little bit more stirring, 50 ml 20% HCl was added SLOWLY - if there's any hydride left, it will bubble vigorously. About 300 mg HgCl2 was added and allowed to dissolve (still stirring) for a couple of minutes. Shredded kitchen aluminum foil was added, around half a gram at a time, allowing the foil to react mostly away before the next portion was added, in order to keep the reaction rate and temperature down. Several grams of Al were used, and the reaction was declared finished when all color was gone and when the chemist thought enough Al had been added. About 500 ml dH2O was added (in a new beaker), and then it was basified with 10% NaOH until the sludge turned from solution -> gel -> granules. This was allowed to settle, then the usual gut-wrenching Al/Hg workup was done, with multiple dilutions, extractions, etc. When the final DCM extractions were vacuumed off, 5.5 ml of impure presumed 2,5-dimethoxyamphetamine freebase (not analyzed, just hoped to be right) was left. Yield: approximately 60%. A drop of freebase produced crystals with the usual ether/IPA/HCl system. This reaction was also run on straight phenyl-beta-nitropropene, except in absolute ethanol and not methanol. Use MeOH! EtOH yields are poor. Anyway, 16g phenyl-beta- nitropropene was reduced to amphetamine freebase, which was then crystallized with 93% H2SO4 in 99% IPA. Yield: 3.5-4g amphetamine sulfate. Bioassaying the product thoroughly kicked the asses of the experimental subjects, and also produced extreme euphoria, most likely due to the joy of having completed a successful synthesis. Osmium Comments: Gonzo's workup is not very good. When he's done with the Al/Hg part he dilutes it with water. The product is still in solution, because the whole mother is still acidic. Then he dilutes it with water, and starts basifying it with NaOH. This precipitates the Al hydroxides and freebases the product at the same time. Bad idea, because the Al sludge will effectively absorb the organic, non-water soluble oil and render it inaccessable for further solvent washes. I'd rather dilute it with some more MeOH, and then start adding the NaOH. This will precipitate the Al just like before, but the MeOH will not be sufficiently diluted to crash out the freebase. It is still soluble in the watery MeOH and filtration will hopefully yield a bigger portion of the product, because the Al sludge will not absorb so much of it. That should give you a better yield, with less solvent washes of the Al hydroxides. Rhodium Comments: The use of Methanol in the reduction of nitrostyrenes is not reccommended, Barium has listed suitable solvents for this reduction in another document. Synthesis of DOB using Urushibara Catalystby Phikshun22,5-dimethoxyphenyl-2-nitropropene10.0g of 2,5 dimethoxybenzaldehyde was dissolved in 50mL of GAA, and 6.8g of nitroethane was added. I dreamed I got both from a photochemical supplier with relative ease. 4.0g of ammonium acetate, that had been stored for weeks over Calcium Chloride (i.e. quite anhydrous), was added and the flask swirled until everything dissolved. This mixture was heated over a pot of boiling water, with heavy duty foil surrounding the flask and top of pot, to keep steam in. It was left for nearly 3 hours, upon which time it had a cherry red colour, and was allowed to cool. After cooling, solvent was stripped off under aspirator vacuum and about 100mL of dH2O was added. The mixture was then extracted with TCE (tetrachloroethylene, brake degreaser, don't ask), and the TCE distilled off under vacuum to afford 12.4g of impure nitropropene, which was recrystallized in 80mL methanol to ~10g, and was recrystallized again from 75mL methanol to a weight of 8.4g. I have no idea why Shulgin only got 6.7g in his 2,5-DMA synth. 2,5-dimethoxyamphetaminePrepared Urushibara catalyst using these instructions. Had to use a cleaned out 4L milk jug instead of a 4L beaker, and I used nuggetized generic Al foil, but otherwise the same method. Activated 15g of the aforementioned catalyst in 580mL of 40% GAA, with 130g of NaCl dissolved, at 70°C for a little over 7 mins. Rinsed with 60 deg C dH2O and washed with IPA, then placed in a 500mL erlenmeyer with 375mL of anhydrous IPA, and 6.7g of the long orange crystalline needles of nitropropene prepped in the previous step. The yellow-orange tint completely leaves the solution in about an hour. For stirring, SWIM used a 12V computer fan, with the blades cut off and a glass stir rod (slightly bent on the end) fastened on with a rubber stopper and some crazy glue. This was attached with a 12V supply with a variable resistor in place to control speed. Worked famously, and totally quiet, which is good, because this dream took place in a small apt. 15g of Al foil prepped in the same fashion as used to precipitate catalyst, is added gram wise, followed by the addition of 3ml 31.45% HCl (HW store grade). This took about 2.5 hours. The flask got quite warm near the middle of the reaction, but never achieves reflux. After Al stops reacting, and H2 is no longer bubbling, 150ml of IPA is added, and then the reaction is basified with 50% NaOH sol. After bubbling subsides, nice yellowish alcoholic layer settled out on top. This was decanted, and concentrated at aspirator vacuum. The remaining AlO sludge is extracted with xylene, and the xylene is added to the other extracts. This was washed once with Brine, and once with dH2O, then dH2O is added, and triturated to a pH of 4. Aqueous extract is kept, basified with NaOH solution and extracted with DCM. DCM is evaporated overnight to provide ~3ml of orangish brown oil. Further A/B extractions of the xylene proved not to contain any more DMA. 4-bromo-2,5-dimethoxyamphetamineThe crude DMA, which weighed 2.1g, and 15ml GAA was placed in a 50ml RBF with a claisen adaptor attached, a condenser, then a gas inlet adaptor, which was hooked to a tube leading outside. Upon the claisen was placed a 125ml addition funnel. Into the funnel was added 2g elemental bromine (I chilled it first in the fridge, still reeked) in 5ml GAA. This was dripped in over a 5 minute period with magnetic stirring, and was left for about 4 hours. This mixture was then added to 200mL dH20, and washed with 3x50mL portions of xylene. This was basified with a 25% NaOH solution, then extracted with 100mL and then 50mL of DCM. Upon evaporation of the solvent, about 3-4ml of a pale brown oil was recovered, which crystallized on standing overnight. To this was added sufficient IPA to dissolve, and it was triturated dropwise with 31% HCl until a pH of 4 was obtained. IPA was distilled off until about 20mL remained, then cold acetone was added to crash out the crystals. Solution was vacuum filtered, and the resulting off-white crystals where recrystallized in IPA to afford 1.25g of perfectly white, fine, powder of DOB.HCl. This worked out to about 415 hits, at 3mg a piece. Personally, I really don’t enjoy DOB that much, but it makes acid look like kids stuff. My roommate loves the stuff though... Synthesis of 2,5-DMA using Urushibara Catalystby ErnyThis is the most successful procedure for the reduction of nitropropenes on Urushibara nickel catalyst I have ever seen in my dreams, and, I guess, the last one: since a long while the only reducing agent I've dreamed about was LAH. 2,5-Dimethoxyamphetamine from 2,5-dimethoxyphenyl-2-nitropropene To a 100 ml rb flask, fitted with a mechanical stirrer and containing 10 g of zinc dust and 3 ml water there was added 3 g of NiCl2x6H2O (of a "chemically pure" grade, IMHO, it is not necessary, purity of zinc is more important), dissolved in 10 ml of water, and the stirring begun. A vigorous reaction took place and the contents of the flask almost ran over; after 15 minutes stirring was stopped and the reaction mass was diluted with tap water and transferred into a 500 ml rb flask, where it was washed several times with tap water and the last time - with distilled water. The precipitated nickel was a dark-gray solid that did not differed much in color from the original zinc dust, when wet. It was digested with 160 ml of 13% acetic acid for 5 min. Zinc dissolution in acetic acid was exothermic and was accompanied by a violent evolution of a hydrogen gas, at the end black catalyst raised to the surface of the solution. All was then filtered with suction on a Schott filter, washed with 200 ml of distilled water and with small amount of IPA. The filtrate had a greenish color. The catalyst was then transferred to the 500 ml rb flask, while still wet, together with 100 ml of IPA. There was then added a solution of 0,2 g of 2,5-DiMeOP2NP in 15 ml of IPA (the resulting solution was VERY diluted, it is not a must), 1 g of thin aluminum foil and 3 ml conc. hydrochloric acid, the mechanical stirrer was fitted to the neck of the flask and the stirring begun. Reflux condenser was not used. Reaction of aluminum with HCl starts slowly but then accelerates, so that the temperature of the reaction mass rises to 60-70°C. When almost all the aluminum had dissolved, another g was added together with 3 ml conc. HCl. All in all, 4 g of aluminum and 12 ml of HCl was added during 2,5 hours. At the end of the reaction enough conc. sodium hydroxide soln. was added to the RM so that the layers could separate. The upper light-yellow IPA layer was decanted, the lower layer washed once with a small amount of IPA, the extracts were combined, and IPA was distilled off. The remaining orange oil was dissolved in petroleum ether from which the amine was acidified out with HCl. This gave 80 mg of 2,5-DMA hydrochloride (~38%) as a light-brown caramel-like substance that did not crystallized at once (it sometimes needs a week to do that, even in a purer state) and was successfully brominated. |