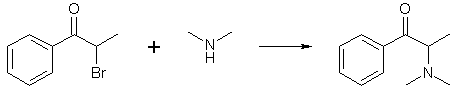
Recently a fellow bee, using information from a reference I sent him/her, carried out a reaction between dimethylamine and α-bromopropiophenone successfully.
I don't have the exact experimental details with me but in general he/she placed the expected amount of dichloromethane in a 300 mL flask and added 35 grams of α-bromopropiophenone (α-BP) to it. The α-BP had a greenish tinge and was added straight from the bottle (purchased rather than made, I guess). The DCM solution immediately took on the same greenish tinge and within seconds, his/her eyes started to water and he/she began to feel incredibly dizzy - this with the hood turned on (but the drap was missing; dummy) while wearing a gas mask.
Into the addition funnel was added the correct amount of 40% aqueous dimethylamine and this was slowly dripped into the stirred DCM/α-BP mixture over a period of about 30 minutes. As the dimethylamine was added, the tear gas effect began to diminish. As indicated from an immersed thermometer, the reaction was exothermic, but not very much so. For about an hour, the biphasic mixture was stirred and generally remained within the 30-35°C range. About an hour after the addition, the temp started to go down and was soon at room temperature where it remained for the next twelve hours.
After the stirring was stopped and the phases separated, the tear gas effect was completely gone, as was the greenish tinge that was clearly evident at the beginning of the reaction. The phases were separated from each other using a sep funnel -- this being done under a hood as the dimethylamine is very smelly and toxic -- and the lower DCM phase was several times with water, the disposal of which was a very smelly task (dimethylamine vapor). Then, the DCM phase was extracted with 10% HCl, which caused its volume to become significantly reduced, and, strangely enough, take on it's previous greenish-tinge character. The DCM phase was separated, and the acid phase was washed once with toluene. Then, stupidly, without cooling, the acid phase was basified with 25% NaOH, which cause the solution to heat up considerably. After cooling down, the aqueous phase separated as a clear solution while a discernable yellow oil floated on top. The whole mixture was extracted with toluene, which was then separated, washed with water (the work-up presenting little trouble as is typical of other syntheses), the toluene dried, and bubbled with HCl gas to yield approximately 25 grams of suspected dimethylcathinone-HCl. This was washed with acetone and dried. A quick sniff indicated the presence of decomposition products -- knowledge of which can only be gleaned from previous experience with the methcathinone synthesis which yields a similar contaminant bearing a similar smell.
Several more washes removed almost all of the by-product, and a sample (about 100 milligrams) was consumed intranasally. There was a slight burning sensation, but not a very distracting one. No obvious effects were noted at this point. Another dose of equal potency was consumed. There was within 30 minutes a slight "speedy" effect, but one that was rather weak. The sensation in the throat and nasal passages was VERY reminiscient of previous experiences with methacathinone consumption, but without any of the immediate intoxicant effects. Notably, there was practically no effect on the blood pressure, nor was the heart-rate increased. In effect, no cardiovascular side effects whatsoever.
Over the next two days, much of the product was consumed; again, there was little in the way of CNS or cardiovascular stimulation but there was a relatively faint pleasant sensation that seemed closer to sedation that anything else, which was very short-lived. Near the end of the experience however (a day or two later), after about two grams had been consumed, there was a marked effect on the blood pressure as well as the presence of occassional facial ticks and increased anxiety and paranoia. But this can hardly be characterized as typical of a normal dose. There was no self-reinforcing effects that in any way could shed light on the drug as having a high addictive profile. In short it was weak and undesireable and thus, there is no interest in repeating the experience. The synthesis was, however, the easiest and cleanest this bee has ever performed; therefore, substituing a methylamine solution for the dimethylamine one could be viable as a way to synthesize methcathinone. The problem of pyrazine contamination could be minimized by the use of DCM as the solvent medium as hydohalogenic solvents allow stabilization of cathinone bases, increasing the yield of desired product and decreasing the amount of by-products.
If the above reaction was to be performed again, it would be best to incorporate into the synthesis the following modifications:
- Use a draped hood.
- Incorporate an enclosed system using a claisen adapter and addition funnel so that the vapors of dimethylamine and more important α-BP, will be contained.
- Before adding the α-BP, cool it in the freezer so that escape of it's vapors will be minimized.
- When performing the basification, cool the solution first and use sodium bicarbonate to neutralize the acid mixture. This will decrease the exothermic nature of the acid neutralization as well as the degree of basicity of the solution, which one of the most significant factors contributing to the dimerization of the cathinone molecule to form the pyrazine contaminant(right behind excess heat).
- Use DCM as the crystallization solvent. During the extraction, the solvent will protect the cathinone base from dimerizing and when crystallized, will probably solvate most of the pyrazine contaminant, making the final product purer.
- If the smell of the pyrazine is still present, recrystallize the final salt. This will remove all of the last traces of by-product.
Addendum on Pyrazines
The contaminant is A "pyrazine", not pyrazine itself. Under basic conditions, (and with heat) cathinones that are mono alkylated condense with another molecule of cathinone to dimerize into the respective pyrazine (my guess is that the carbonyl groups react with the other molecules amino group to bring about this change). For more information about cathinone-pyrazine formation refer to the article by Bertold D. Berrang et al.1, in which the stability of cathinones in various solvents is reviewed, plus the synthesis of cathinone itself from norpseudoephedrine. (Interestingly and in total contrast to the information above, you should know that cathinone salts are very stable). More information detailing the formation of the pyrazine has been given by M. M. Lee2.
Regarding the pyrazine toxicity, I cannot speak for any other than that produced by dimerization of methcathinone base. This is a problem worthy of Dal Cason or some other researcher and should be tackled ASAP. As for the pyrazine, it has a fishy unpleasant smell that first manifests itself as white crystalline compound that later, after several days, darkens to a yellow color. It can be washed out with acetone and preferably DCM, but my guess is that anything short of crystallization will not remove all of it. It causes in almost everyone that I have observed a marked rise in blood pressure, but for most, this effect is hardly noticable. Curiously, when taken without the presence of methcathinone (or at least, very little of it) one's pulse stays at a low pace, while the blood pressure increases significantly. For me, this symptom is quite pronounced; for others, I have yet to notice too much of a chronic trend. However, everyone who injests this substance takes on a sunken eye appearance and just plain looks sick. And in total contrast, pure methcathinone can be ingested for almost two days straight without any significant side effects (thus, in my opinion, proving that methcathinone that has been properly washed of all or most impurities is relatively safe). Additionally, the presence of the pyrazine contaminant magnifies the paranoid aspect to the experience.
Fester, in the last edition of his book "Secrets of Methamphetamine Manufacture" without realizing it, alludes to the pyrazine in his Methcathinone chapter as a contaminant which he was not familiar with but which he knew was dangerous to take intraveneously as it causes the coagulation of one's blood. This is just a guess but based on the way it feels, that description would be accurate; however, I do no shoot up drugs and have still suffered this effect intranasally.
Removal of this contaminant is easy and should not be skipped. Although it takes quite a while to notice it's effect on the methcathinone experience, eventually with extended use, all of the negative effects that I mentioned above will no doubt take their course.
As for the potential fatality of using such a contaminated product? Once - as a separate, isolated product (as in not mixed with actual methcathinone) - ingestion of it almost killed me, but my hands are tired and I'll tell that story later. Needless to say, it was not typical and downright stupid and is both proof that ingestion of methcathinone can be both safe AND dangerous, depending on the conditions... more on this later.
Experimental
o-Methoxy-α-bromopropiophone3
One hundred and forty-seven grams (0.895 mol) of o-methoxy-propiophenone was dissolved in 500 ml of chloroform and with stirring 144 g. (0.9 mol) of bromine in 250 ml of chloroform was added over a period of 1.5 hours with cooling to 20°C. The solution was allowed to stir an additional 2 hours at room temp, after which air was bubbled through it for 30 minutes. The amber solution was washed with water, sodium bicarbonate, then water, and dried. The pale green yellow solution was stored in the refrigerator until use. The yield was about 80%. The bromoketone could be used in this form for the next step, but somewhat better yields were obtained if the chloroform was removed, the oil dissolved in toluene which was then chilled in Dry Ice and petroleum ether added. On scratching, the bromoketone came out as pale green crystals melting below 40°C. yield about 50%.
o-Methoxy-alpha-methylaminopropiophenone
Without any further purification the above crystalline bromoketone (45.5 g;0.187 mol)in 150 ml of chlorform was stirred vigorously at 35-40°C and a solution of 15 g. (0.48 mol) of methylamine in 60 ml of water was added dropwise over a 30-min period. Stirring was continued an additional 1.5 hours at that temp. The chloroform layer was washed three times with water, dried and the solvent removed under vacuum. The residue was dissolved in ether which was poured into cold ethereal hydrogen chloride. The resulting yellowish gun soon set to a solid. Two recrystallizations from absolute alcohol gave 8.5 g. of beautiful long white needles, mp 175-177°C. The filtrates yielded an additional 9 g of white crystals; total yield 41%.
BTW, in the same paper3 is an excellent Leuckart reaction scheme where they use ammonium hydroxide and formic acid directly in the soup instead of formamide. Also details synthesis of the nitroalkene and it's reduction to P2P using a biphasic mixture in a "through process" fashion.