Summary
Four deuterium-labelled compounds were prepared for use as internal standards in the quantification of methamphetamine, DOM, PCP, and methaqualone at low levels in body fluids by selected ion monitoring. The need for standards containing more than three deuterium atoms per molecule and having high isotopic purity is discussed.
Introduction
The technique of selected ion monitoring1, which was originally given the name mass fragmentography2, has proven extremely useful in recent years for the quantification of organic compounds at low levels. A number of recent reviews3-6 describe this technique in detail and summarize the application of selected ion monitoring to the qualitative and quantitative determination of drugs, pesticides, and food additives in complex mixtures.
Quantification by selected ion monitoring may involve the use of internal standards. In the ideal situation, the standard differs only in mass, and is identical in all other physical and chemical properties. A strong case has been made for the use of compounds labelled with stable isotopes7,8. Deuterium is the most accessible and the least expensive stable isotope for use in preparing internal standards, although 15N, 13C, 18O, and others have also been used.
There are several ways that deuterium can be introduced, perhaps the easiest of which is to use derivatizing agents containing deuterium. For example, bis(trimethylsilyl)acetamide-d18 has been used to introduce trimethylsilyl-d9 groups9. Methoxylamine-d3 has been used to form a methoxime-d3 derivative from a carbonyl function10, and an O-methyl-d3 derivative of a phenol has been prepared using methyl-d3 iodide11. The compound being determined must of course be derivatized with the undeuterated derivatizing agent prior to quantification.
The advantage of using derivatizing agents containing the stable isotope is that derivatives are easily prepared and therefore a large number of different standards can be quickly obtained. The major disadvantage of this technique is that derivatization cannot take place until after much of the sample manipulation has been completed. It would be much better if the internal standard were to go through the entire isolation and analysis procedure. In this way, losses could be compensated and the internal standard could act as a carrier for very low levels of the compound being determined.
Since our objective is the quantitative determination of drugs at very low levels, we felt it necessary to prepare labelled compounds in which the deuterium atoms were incorporated into the parent molecule thus overcoming the disadvantages of compounds labelled by derivatization.
The next consideration was the number of deuterium atoms to be introduced per molecule of standard. Standards with too few deuterium atoms (one or two) are likely to be contaminated by undeuterated compound, and in addition, any fragmentation involving loss of hydrogen or deuterium is likely to contribute to the mass of the undeuterated compound being monitored. In these cases, the standard will introduce a background making quantification more difficult at low drug levels or at high standard/drug ratios. On the other hand, too many deuterium atoms will make the standard significantly different from the drug. Even with three deuterium atoms per molecule in compounds of modest molecular weight, a slight separation of standard and drug can be observed on a GC column; and the object is to keep the standard as similar to the drug as possible.
However, in chemical-ionization (CI) selected ion monitoring, even three deuterium atoms per molecule may not be sufficient. A common ion in CI-MS corresponds to (M+1)+, where M is the mass of the parent compound. The corresponding peak for the standard containing three deuterium atoms per molecule would be at (M+3+1)+. However, the peak of a standard corresponding to loss of deuterium, (M+3-2)+ would correspond to the (M+1)+ ion of the parent drug and would constitute an interference from the standard. This interference has been observed in several cases in our laboratory.
A reasonable compromise was to prepare standards with at least 4 deuterium atoms per molecule but with as few in excess of 4 as was convenient from a synthesis standpoint. Using this guideline, for the reasons already stated, synthesis of four deuterium-labelled standards was undertaken.
Chemistry
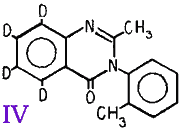
We describe here the synthesis of N-(methyl-d3)-α-methylphenethylamine-α,β-d2 (methamphetamine-d5) (I); 2,5-di(methoxy-d3)-4-methylamphetamine (DOM-d6) (II); 1-[1-(phenyl-d5)cyclohexyl]piperidine (PCP-d5) (III); and 2-methyl-3-o-tolyl-4(3H)-quinazolinone-5,6,7,8-d4 (methaqualone-d4) (IV) for use as internal standards in the determination of methamphetamine, DOM, PCP, and methaqualone, respectively. Each standard contained from 4 to 6 deuterium atoms per molecule and was of high isotopic purity.


Methamphetamine-d5 (I) was prepared by a combination of procedures given in the literature. α-Methylphenethylamine-α,β-d2 (amphetamine-d2) was prepared using a slight modification of the procedure of Lindeke and Cho12 from 1-phenyl-2-nitro-1-propane. An N-trideuteromethyl group was then introduced using the general procedure suggested by Marshall and McMahon13 in which the N-carbethoxy derivative of amphetamine-d2 was formed and reduced to methamphetamine-d5 with lithium aluminum deuteride.

DOM-d6 (II) was prepared by reacting methylhydroquinone with dimethyl-d6 sulfate to form 2,5-di(methoxy-d3)toluene, which was converted to 2,5-di(methoxy-d3)-4-methylbenzaldehyde. The benzaldehyde derivative was condensed with nitroethane to form 2-nitro-1-[2,5-di(methoxy-d3)-4-methylphenyl]-1-propene, which was reduced with lithium aluminum hydride to DOM-d6.
PCP-d5 (III) was prepared by reacting 1-piperidinocyclohexanecarbonitrile with the Grignard reagent from bromobenzene-d5 after the method of Kalir, et al., that was used for the synthesis of unlabelled PCP14.
Methaqualone-d4 (IV) was prepared from phthalimide-d4. Anthranilic-d4 acid was prepared by the Hofmann reaction of phthalimide-d4 and was then converted to N-acetylanthranilic-d4 acid which was condensed with o-toluidine to form methaqualone-d4.
Experimental
N-(Methyl-d3)-α-methylphenethylamine-α,β-d2 (Methamphetamine-d5) (I)
α-Methylphenethylamine-α,β-d212
To 2.2 g (52 mmole) of lithium aluminum deuteride in 100 ml of anhydrous ether was added 3.3 g (20 mmole) of 1-phenyl-2-nitro-1-propene15, dissolved in 100 ml of ether, dropwise at such a rate as to maintain reflux (30 min for addition). The solution was then refluxed 3 hours, cooled in an ice bath and 5 ml of water added dropwise with caution. Then 100 ml of a solution con taining 30 g of Rochelle salt was added. The ether layer was separated and the aqueous layer extracted twice with ether. The ether solutions were combined, dried over anhydrous potassium carbonate, filtered, and concentrated. The crude product was distilled and the fraction boiling 89-99?C/10mmHg was collected. The yield was 1.8 g (64%).
N-Carbethoxy-α-methylphenethylamine-α,β-d2
To a solution of 1.5 g (11 mmole) of α-methylphenethylamine-α,β-d2 in 10 ml of chloroform was added 1.0 g of sodium hydroxide in 5 ml of water followed by 1.8 g (16.6 mmole) of ethyl chloroformate, and the mixture was stirred vigorously for 20 min. The chloroform solution was separated and the aqueous layer extracted twice with 5-ml portions of chloroform. The chloroform solutions were combined, washed with 6N hydrochloric acid, then with water, dried (MgSO4), filtered, and the solvent removed on a rotary evaporator. The clear oil would not crystallize; by GC it was >98% pure. The yield was 2.2 g (96%). When the same reaction was run on optically active amphetamine, the N-carbethoxy derivative formed in this way had mp 47-48.5?C, in good agreement with the literature16. Apparently the racemate has a much lower melting point.
N-(Methyl-d3)-α-methylphenethylamine-α,β-d2 (I)
To 1.0 g (24 mmole) of lithium aluminum deuteride in 40 ml of tetrahydrofuran (THF) was added 2.0 g (9.6 mmole) of N-carbethoxy-α-methylphenethylamine-α,β-d2 in 10 ml of THF over 20 min at room temperature. The resulting mixture was refluxed 2 hours. With ice-bath cooling, 3.5 ml of water was added dropwise with caution, and the resulting mixture was stirred 30 min at room temperature. The aluminum salts were filtered and washed twice with hot THF. The filtrate and washings were combined, dried (MgSO4), filtered, and concentrated to about 10 ml on the rotary evaporator. The crude product was distilled and the fraction boiling 91-92?C/10 mmHg collected. The yield was 0.95 g (65%). A portion of the product was converted to the hydrochloride with a saturated solution of hydrogen chloride in ether. The mp of the salt was 133-134?C (lit.17 mp for the undeuterated racemate 133-135?C).
2,5-Di(methoxy-d3)-4-methylamphetamine (DOM-d6) (II)
2,5-Di(methoxy-d3)toluene
This procedure is an adaptation of that used by G.N. Vyas and N.M. Shah for the formation of the dimethyl ether of quinacetophenone18.
To a hot solution of 9 g (73 mmole) of methylhydroquinone in 60 ml of ethanol under nitrogen, a solution of 7.5 g (187 mmole) of sodium hydroxide in 20 ml of water and 20 g of dimethyl-d6 sulfate were added alternately in approx. 3-ml portions starting with the sodium hydroxide solution. After addition, the solution was refluxed 1 hour and then 2 g of sodium hydroxide in 5 ml of water was added and the solution refluxed an additional hour. The ethanol was removed on the rotary evaporator and the residue was taken up with water and extracted three times with ether. The ether extracts were combined, washed with water, dried (MgSO4), filtered, and concentrated. The crude product was distilled and the fraction boiling 110-113?C/20 mmHg collected. The yield was 8.5 g (74%). GC showed the product to be 95% pure with the monomethyl ether (identified by GC-MS) as the only impurity.
2,5-Di(methoxy-d3)-4-methylbenzaldehyde
The undeuterated aldehyde has been prepared by the Gattermann reaction19 and is reported in the literature20. To an ice-cooled solution of 8 g (51 mmole) of 2,5-di(methoxy-d3)toluene in 30 ml of benzene was added 5.0 g (185 mmole) of dry hydrogen cyanide21 followed by 11 g (83 mmole) of aluminum chloride. After hydrogen chloride had been passed through the mixture for 4 hours at 35?C (oil bath), it was poured into a mixture of concentrated hydrochloric acid (25 ml) and ice (200 g). The benzene and product were then steam distilled - approx. 1200 ml of distillate was collected. The product was taken up in ether and the ether solution washed once with water, dried (MgSO4), filtered, and the solvent removed on the rotary evaporator. The crude aldehyde, 7.6 g (80%), was crystallized from ethanol to give 5.9 g (62%) of white crystals, mp 83-84?C (lit.20 mp for the undeuterated aldehyde 84-85?C).
2-Nitro-1-[2,5-di(methoxy-d3)-4-methylphenyl]-1-propene
To 20 ml of glacial acetic acid in a 50 mL round-bottomed flask was added 5.0 g (27 mmoles) of 2,5-di(methoxy-d3)-4-methylbenzaldehyde, 2.7 ml (38 mmole) of nitroethane, and 2.0 g of ammonium acetate. The solution was refluxed 3 hours, cooled, and poured into ice and water. After the ice had melted, the yellow solid was washed well with water and pressed dry. Crystallization from about 40 ml of methanol gave 2.8 g (43%) of a bright-yellow, microcrystalline solid, mp 90-91?C (lit.22) mp for the undeuterated compound 85.5-87.5?C).
2,5-Di(methoxy-d3)-4-methylamphetamine (DOM-d6) (II)
To 1.2 g (31 mmole) of lithium aluminum hydride in 75 ml of anhydrous ether was added 2.2 g (9 mmole) of 2-nitro-1-[2,5-di(methoxy-d3)-4-methylphenyl]-1-propene in 50 ml of ether dropwise at such a rate as to maintain reflux. The solution was then refluxed 3 hours, cooled in an ice bath, and 10 ml of water added dropwise with caution followed by 75 ml of a 30% Rochelle salt solution. The ether layer was separated and the aqueous layer extracted twice with ether. The ether solutions were combined, washed with water, dried (MgSO4), filtered, and the solvent removed on the rotary evaporator. The residue, 1.75 g, was crystallized from hexane in the cold to give 1.3 g (67%) of white crystals with mp 58-60?C. The free base was converted to the hydrochloride with a hydrogen chloride-saturated ether solution. The precipitate was crystallized from ethanol/ether to give white crystals, mp 186-188?C (lit.23) mp for undeuterated DOM·HCl 190?C, and an authentic sample had mp 187-189?C).
1-[1-(Phenyl-d5)cyclohexyl]piperidine (PCP-d5) (III)
Phenyl-d5 magnesium bromide was prepared from 10.0 g (62 mmoles) of bromobenzene-d5 and 1.6 g (67 mmoles) of magnesium metal in 75 ml of ether. To the Grignard reagent was added 10 g (52 mmole) of 1-piperidinocyclohexanecarbonitrile in ether-benzene (25 ml/10 ml) over 30 minutes. After the reaction mixture had stirred 3 hours, it was cooled with an ice bath and 75 ml of a saturated solution of ammonium chloride added. The ether-benzene solution was separated and washed with water. The product was extracted twice with 6N hydrochloric acid (50 ml portions). With ice-cooling, the combined acid extracts were made basic with concentrated ammonium hydroxide (50 ml). The product was extracted into ether and the ether solution washed twice with water, dried (MgSO4), filtered, and the solvent evaporated. The residue was distilled and the fraction boiling 120-124?C/0.35 mm was collected. The yield was 4.8 g (37%). A forerun of 4.6 g (53%) consisting almost entirely of 1-piperidino-1-cyclohexene was also obtained. This probably results from elimination of HCN from the starting material by action of the Grignard reagent. The free base was converted to the hydrochloride, which was crystallized from ether-ethanol. The hydrochloride had mp 219-220?C, the same as that of an authentic sample of the undeuterated hydrochloride.
2-Methyl-3-o-tolyl-4(3H)-quinazolinone-5,6,7,8-d4 (Methaqualone-d4) (IV)
Anthranilic-3,4,5,6-d4 acid
This preparation is based on the formation of anthranilic acid from phthalimide as described by Weygand24 To a cooled solution of 10 g of sodium hydroxide in 50 ml of water was added 8.0 g of bromine. The resulting solution was cooled to -10?C and 7.5 g (50 mmole) of phthalimide-3,4,5,6-d425 added as a slurry in 10 ml of water over about 15 min with strong stirring. The freezing mixture was removed and the reaction mixture allowed to slowly warm to 10?C (about 30 min), at which point all the solid had dissolved. Pulverized sodium hydroxide (6 g) was then added and the temperature rose to 45?C. The solution was heated to 80?C, cooled, and then neutralized with 16 ml of concentrated hydrochloric acid and acidified with 8 ml of acetic acid. After standing in the cold, the solid was filtered, washed with cold water, and dried. The yield was 4.85 g (69%), mp 144-145?C.
N-Acetylanthranilic-3,4,5,6-d4 acid
To 3.3 g (23 mmole) of anthranilic-3,4,5,6-d4 acid was added 10 ml of acetic anhydride, and the mixture was refluxed 1 hour. The solution was then cooled, 3.5 ml of water added, and the solution again brought to reflux. Ten ml more of water was added at reflux and then the solution allowed to cool. The resulting crystals were collected and washed several times with water. The yield was 3.9 g (91%), mp 183-186?C26.
2-Methyl-3-o-tolyl-4(3H)-quinazolinone-5,6,7,8-d4 (Methaqualone-d4) (IV)
Different methods were tried for the condensation of N-acetylanthranilic acid and o-toluidine, based on several literature sources27-30. Following is a description of the procedure that was used.
To 3.0 g (16.4 mmole) of N-acetylanthranilic-3,4,5,6-d4 acid in 10 ml of toluene was added 1.8 g (16.8 mmole) of o-toluidine. To this mixture was added 3.0 g (19.6 mmole) of phosphorus oxychloride in 10 ml of toluene dropwise over 15 min. The mixture was refluxed 2 hours with mechanical stirring. Toward the end of the reaction stirring became difficult because of the formation of a sticky solid. The reaction mixture was cooled and poured into 100 ml of a 10% solution of sodium carbonate. The solid was washed into the sodium carbonate solution with methanol. The methanol/toluene were removed by steam distillation (boiled off) and after the remaining oil had crystallized it was filtered off, washed with water, taken up in hot methanol, treated with activated charcoal, and the solution filtered. To the boiling filtrate was added water until the solution became turbid, and the product was allowed to crystallize. The light-yellow product was filtered, washed with water, and dried. The yield was 2.5 g (60%), mp 111-114?C. The yellow color can be removed by chromatography on basic alumina using chloroform as eluent. The product comes off rather quickly while the yellow impurity moves more slowly and is retained as a sharp band. The amount of this impurity and the yield of the reaction seems to depend on the purity of the starting N-acetylanthranilic acid. The chromatographed product was then crystallized from hexane/methanol to give white crystals, mp 115-116?C (lit.28) mp for the un deuterated compound 115-116?C).