Abstract
Rate of copper(I)-catalyzed substitution of aryl bromides into methyl-aryl ethers by methoxide ion is greatly enhanced in presence of esters. Soluble copper(I) complexes 2, arising from tetrahedral adducts 1 of methoxide onto esters. are likely responsible for this improvement.

Copper-catalyzed reaction of sodium methoxide with aryl bromides in methanol is generally a method of value for the preparation of methyl aryl ethers1:
However, this process is often sluggish or even ineffective in the case of unactivated (devoid of electron withdrawing substituents Z) aryl bromides such as bromobenzene, and various studies have been undertaken to resolve this problem. Several publications and patents claimed that amide co-solvents such as N,N-dimethylformamide (DMF) were very useful2a-d, allowing satisfactory solubility and stability of cuprous salts catalysts, which are less soluble and readily disproportionate in methanol alone. Remaining disadvantage was the high cost of such solvents face to methanol, and R. J. Bryant reported that formamides could be replaced by alkyl formates3, in relatively small proportions toward methanol solvent. Methoxylation of bromobenzene so proceeds in 56% yield and 2-bromophenol is quantitatively converted into guaiacol.
More recently, H. L. Aalten et al. revisited this chemistry, concluding inter alia that amide co-solvents (DMF) were quite effective to obtain a quantitative bromobenzene → anisole substitution, but methyl formate was curiously found to have no effect on reaction rate in methanol4.
Aiming at a better understanding of this reaction, we firstly checked that methyl formate really improves the rate of substitution in Bryant's conditions, i. e. with sodium methoxide concentrations higher than 3M, but not with a 1.6M solution as used by H. L. Aalten et al.4 At this stage, it seemed not substantiated to assign to the sole -CO-H formyl group a specific activity unshared by others -CO-R substituted groups; at the contrary, we report here that any (stable) ester of general formula R'-O-CO-R is able to co-catalyze methanolysis of aryl bromides.
Kinetic follow-up of the transformation of 2-bromophenol into guaiacol with various added esters evidences a great improvement in rate referred to the sluggish classical reaction, as shown by the following graph:

Transformation of 2-bromophenol:
C0 = initial concentration, C = concentration at time t.
Plot 1: no ester added (blank); 2: methyl formate;
3: methyl benzoate; 4: ethyl oxalate; 5: diethyl carbonate
and dimethyl maleate (superimposed); 6: ethyl acetate.
Esters (3 mmol) are added to 5M NaOMe solution in MeOH
(6 ml) under argon, then 2-bromophenol (5 mmol) and CuBr
(1 mmol) and the mixture is stirred and heated to reflux.
First order-like plots, due to the large excess of methoxide, are obtained in each case, except with methyl benzoate (Plot 3) where metallic copper appeared after 1 hour (decomposition of catalyst). Slope ratio between the blank experiment (Plot 1) and the ethyl acetate one (Plot 6) is superior to 20, demonstrating the effectiveness of this co-catalyst (completion is reached within 45 min., with a quasi-quantitative yield).
Use of ethyl rather than methyl esters leads sometimes to traces of 2-ethoxy phenol (0.5 to 1%), due to releasing of ethoxide ion through transesterification of the co-catalyst by methoxide; methyl esters may therefore be preferred if highest purity of final product is required.
Anisole is also quasi-quantitatively obtained from bromobenzene in presence of ethyl acetate (10 mmole PhBr, 4 mmol EtOAc, 1,4 mmol CuBr in 6 ml 5M MeONa/MeOH, 98% conversion after 1h reflux, 100% after 2h, yield of isolated anisole >95% without detected phenetole).
Another example, of industrial interest, is the transformation of 5-bromovanillin into syringaldehyde: substrate (5 mmol) is refluxed with EtOAc (3 mmol) and CuBr (1 mmol) in 5 M MeONa/MeOH (10 ml) for 14 h; classical work-up leads to pure syringaldehyde (95%). When starting from more soluble 5-bromovanillin dimethyl acetal, reaction is achieved within two hours (yield 98%).
Hence, the positive influence of esters on the course of these aromatic copper-catalyzed substitutions is well established, and the necessity of high methoxide concentration is checked and explained by the following findings:
Whereas copper(I) bromide (or chloride) catalyst, once added to methoxide solution, gives rise to insoluble yellow copper(I) methoxide, which decomposes under refluxing to Cu0 and Cu(II), presence of an ester in concentrated (3 to 5 M) methoxide solutions prevents precipitation of copper(I) methoxide, but provides a colorless mixture in which added aryl bromides are readily transformed into methyl aryl ethers. Use of smaller amounts of copper (0.5 mmol CuBr in 15 ml 4M MeONa/MeOH) even leads to colorless clear solutions (copper complex and NaBr are entirely soluble under MeOH refluxing). Lower methoxide concentrations (up to 2.5 M) render esters co-catalysts ineffective, as copper(I) methoxide remains insoluble; further addition of methoxide to the mixture leads again to clear solutions.
High efficiency of ethyl acetate is not due to any base-catalyzed condensation into ethyl acetoacetate, since the latter has a negative effect on the reaction: it slows the rate down to ~75% of the blank's one when added to the 2-bromophenol / CuBr / MeONa system. The observed stabilization and increased solubility of Cu(I) catalyst in presence of esters MeO-CO-R - even non-enolizable - can then be attributed to their adduct with methoxide ion, obtained at high concentrations of the latter, and written as follow:
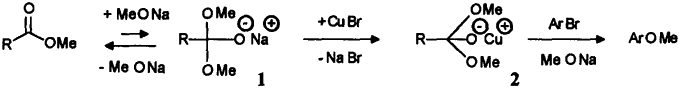
Tetrahedral intermediate 1 is well known to be formed in very small amounts during base-catalyzed methanolysis of esters;5a,b it is obviously here a potent ligand for added copper(I), tentatively drawn as complex 2, allowing in turn the substitution ArBr → ArOMe to proceed with a great efficiency. Such a stabilization of primary catalyst, and also probably of various Cu(II) or Cu(III) intermediate species previously proposed to occur in the aromatic substitution6 itself, implies ester to be substantially transformed (CuOMe no more precipitates from 5M MeONa solution). Such a displacement of equilibrium has yet been described, due this time to favored sp3 hybridization, in the case of trifluoroacetate (R = CF3)7a,b where 1 was the only species detected in dibutylether in presence of MeONa. When used in the same conditions than other esters, methyl trifluoroacetate is readily decomposed in concentrated and/or heated MeONa solutions8 (→ CHF3 + MeO-CO-OMe) and the observed catalysis must in fact be attributed to resulting dimethylcarbonate. On the contrary, CF3COOMe allows formation of clear colorless solutions of copper(I) at 20°C and quite lower methoxide concentrations (e.g. 10 mmol CF3COOMe and 16 mmol MeONa in 13 ml MeOH, then 0.5 mmol CuBr), and thus brings confirmation to the above hypothesis of a tetrahedral chelating intermediate 2 to be the key of ester co-catalysis; due to enhanced tendency of fluorinated esters to add nucleophiles, formation of 1 then 2 no more requires high methoxide concentrations. However, despite high solubility of copper(I), aryl bromides (bromobenzene, 2-bromophenol) remain practically unchanged after 2h when submitted to this system: temperature and MeONa concentration are too low to insure substitution step itself. Dibutylether solvent (or dioxane) does not allow that kind of experiment, as CuBr remains insoluble at 20°C, while metallic copper readily forms on heating; on the other hand, strongly absorbing MeONa/MeOH solutions prohibits to perform infrared studies on 2 as it was achieved in dibutylether7a to evidence trifluoroacetate adduct 1. Further effort, aiming to characterize or even isolate copper complex 2 , is currently underway.
This work was partly performed with financial support from Rhone-Poulenc Chimie, and industrial applications are patented9.