Introduction
There are many procedures out there for the production of N-methyl-amphetamines (methamphetamines) from various starting materials, such as phenyl-2-propanone (P2P) or ephedrine, but what if you already have an amphetamine (or phenethylamine) and wanted to add a methyl group to the nitrogen atom? If you would use the first reaction that comes to mind for the conversion, to alkylate the amphetamine with methyl iodide or dimethylsulfate, you would be disappointed, as you would get a mixture of products, most important the N,N-dimethyl-amphetamine (of very low activity), as once the amphetamine has been methylated to methamphetamine, the molecule is much more succeptible to another alkylation, and thus the dimethyl- amphetamine is formed much faster than the remaining amphetamine is alkylated to methamphetamine. Actually, in the reaction mix you would find unreacted amphetamine, N-methylamphetamine, N,N-dimethyl- amphetamine and even some of a quaternary N,N,N-trimethylamphetammonium salt.
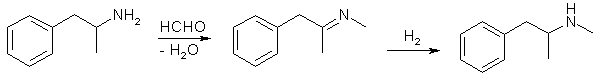
To avoid this happening, we must usually resort to indirect methods of introducing the methyl group. One way is to react the amphetamine with formaldehyde (either as an aqueous solution, or as paraformaldehyde) to get the amphetamine formaldehyde imine, which can then be reduced to the N-methylamphetamine using a several different reducing agents, for example Al/Hg or Pt/H2.
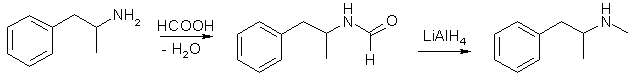
Amphetamine can also from an amide with formic acid, N-formylamphetamine, if boiled in a Dean-Stark apparatus where the formed water in the reaction is continously removed, thus driving the formation of the amide forward. The amide can then be reduced by for example lithium aluminum hydride.
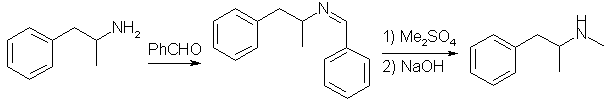
Another way is to react the amine with benzaldehyde to form an imine, which then can safely be alkylated with methyl iodide or dimethyl sulfate, and after hydrolysis of the resulting compound, N-Methylamphetamine is formed.
Finally, it must be said that using exotic reagents like cesium salts, it might be possible to directly mono-methylate amphetamine without too much overalkylating taking place. You get an approximate 9:1 ratio of secondary amine to tertiary amine, while not perfect it is pretty good for one step with no reduction necessary. The selectivity might be somewhat lower with methylations than with the butylation described in the last synthesis of this document.
Formaldehyde and Aluminum Amalgam1
N-Methyl-amphetamines were prepared by the reaction of the corresponding amphetamine with formaldehyde and reduction in the absence of acid. Thus, a mixture of 1 mole amphetamine freebase (136g) and 1 mole aqueous formaldehyde (81ml 37% or 75ml 40%) in 350 ml alcohol and an excess of aluminum amalgam was reduced for several hours, water added, aluminum hydroxide filtered off, the solution acidified and evaporated, and the freebase separated by means of alkali yielding phenyl-N-methylisopropylamine, converted to the hydrochloride, mp 140°C. Similarly, 70g d-amphetamine in alcohol with aluminum and 1 mole formaldehyde gave d-methamphetamine, which was converted to the phosphate salt. 1-Phenyl-2-aminopropanol similarly yielded ephedrine.
Paraformaldehyde, Platinum Oxide and Hydrogen2
This procedure is for the methylation of 2-phenethylamine, but it can easily be adapted for use with any amphetamine.
A solution of 30.25 g (0.25 mole) of 2-phenylethylamine and 7.5 g. (0.25 mole) of paraformaldehyde (Note 1) in 50 ml of 95% ethanol was allowed to stand for a short period (Note 2). Additional ethanol (100 ml), 15 g. (0.25 mole) of glacial acetic acid (Note 3), and 0.5 g. of platinum oxide (Note 4) were added, and hydrogenation was carried out at room temperature and 3 atm. When hydrogen absorption was complete, the catalyst was removed, and the filtrate and washings concentrated to dryness. The residue was treated with sodium hydroxide solution (Note 5), extracted with ether, dried and distilled to give an 80-90% yield of N-methyl 2-phenethylamine (Note 6).
Notes:
- Originally about 20 ml of 37% formaldehyde solution was added (0.25 mole). In view of the reported danger of dimethylation of primary amines when formaldehyde is used, it appeared safer to use paraformaldehyde, since an exact amount could be weighed.
- When the aldehyde and base were mixed, the solution became warm. The solution was allowed to stand for 0.25-0.5 hr. for complete reaction. In some cases the mixture was allowed to stand overnight, but this gave no improvement in yield after reduction. When no heat developed in the reaction of aldehyde and amine, the mixture was warmed slightly and allowed to stand before reduction. When low boiling components are to be mixed, it is best to cool the solution of amine while the aldehyde is added to prevent loss of amine or aldehyde from the exothermic reaction. After standing, the mixture can then be hydrogenated. Hydrogen uptake was usually complete within 2 hr or less.
- The reaction rate in this reduction was slower in the absence of acetic acid. Other acids should also be satisfactory as long a they do not cause reversal of the formation of imine. Strong acids should not be used with rhodium because they act as inhibitors of its activity. Many reductive alkylations were carried out successfully with platinum or palladium in the absence of acid. When reduction was too slow, interruption for addition of the required amount of acid did speed hydrogen uptake.
- Palladium on carbon was not used in this experiment, but 5 g of 5% rhodium on carbon did prove satisfactory. Freifelder states there is little doubt that 5 g of 5% palladium on carbon or an equal amount of 5% platinum on carbon would work as they had in other reductive alkylations.
- When acid is not used, addition of alkali and extraction of base are unnecessary.
- A reaction such as this is actually a reduction of the imine in situ, a very useful procedure that may be used in alkylations with other aldehydes.
Formic acid and Lithium Aluminum Hydride3
A solution of 6.55 g of 3,4-methylenedioxyamphetamine (MDA) as the free base and 2.8 mL formic acid in 150 mL benzene was held at reflux under a Dean Stark trap until no further H2O was generated (about 20 h was sufficient, and 1.4 mL H2O was collected). Removal of the solvent gave an 8.8 g of an amber oil which was dissolved in 100 mL CH2Cl2, washed first with dilute HCl, then with dilute NaOH, and finally once again with dilute acid. The solvent was removed under vacuum giving 7.7 g of an amber oil that, on standing, formed crystals of N-formyl-3,4-methylenedioxyamphetamine. An alternate process for the synthesis of this amide involved holding at reflux for 16 h a solution of 10g of MDA as the free base in 20 mL fresh ethyl formate. Removal of the volatiles yielded an oil that set up to white crystals, weighing 7.8 g.
A solution of 7.7 g N-formyl-3,4-methylenedioxyamphetamine in 25 mL anhydrous THF was added dropwise to a well stirred and refluxing solution of 7.4 g LAH in 600 mL anhydrous THF under an inert atmosphere. The reaction mixture was held at reflux for 4 days. After being brought to room temperature, the excess hydride was destroyed with 7.4 mL H2O in an equal volume of THF, followed by 7.4 mL of 15% NaOH and then another 22 mL H2O. The solids were removed by filtration, and the filter cake washed with additional THF. The combined filtrate and washes were stripped of solvent under vacuum, and the residue dissolved in 200 mL CH2Cl2. This solution was extracted with 3x100 mL dilute HCl, and these extracts pooled and made basic with 25% NaOH. Extraction with 3x75 mL CH2Cl2 removed the product, and the pooled extracts were stripped of solvent under vacuum. There was obtained 6.5 g of a nearly white residue which was distilled at 100-110°C at 0.4 mm/Hg to give 5.0 g of a colorless oil. This was dissolved in 25 mL IPA, neutralized with concentrated HCl, followed by the addition of sufficient anhydrous Et2O to produce a lasting turbidity. On continued stirring, there was the deposition of fine white crystals of 3,4-methylenedioxy-N-methylamphetamine hydrochloride (MDMA) which were removed by filtration, washed with Et2O, and air dried, giving a final weight of 4.8 g.
Benzaldehyde and Dimethylsulfate4
p-Methoxyphenethylamine, generated from 100 g (0.536 mole) of the hydrochloride by stirring with conc. aq NaOH, was treated with 100 ml of benzene (toluene could be substituted) and 70 g (0.66 mole) of benzaldehyde. A mildly exothermic reaction began at once. The mixt was heated under reflux until no more H2O was present in the condensate (ca. 1 hr), then, without cooling, an attached Dean-Stark trap was removed and a soln of 82 g (0.65 mole) of Me2SO45 in 200 ml of benzene (toluene could be substituted) was added through the condenser at such a rate as to maintain reflux (15 min). The 2-phase mixt was heated for 90 min on the steam bath, cooled slightly, treated with 200 ml of H2O, and heated for an addn 20 min. After cooling in ice, the aqueous layer was washed twice with Et2O to remove unreacted benzaldehyde and made strongly basic with 50% aq NaOH. Two Et2O exts of the basic aqueous phase were added to the amine layer which separated, and the resulting solution was evacuated at the aspirator for 30 min, leaving 90 g (102%) of crude N-methyl-p-methoxyphenethylamine. This material was dissolved in 500 ml of 20% abs EtOH-Et2O and treated with 50 ml of conc HCl with swirling and cooling to yield the white, cryst hydrochloride, which was washed thoroughly with ice-cold 20% EtOH-Et2O and dried, mp 185.5-186.5°C. The yield was 83 g (77%).
Cesium Hydroxide Promoted Monoalkylation6,7
Selective N-alkylation of primary amines was developed using cesium hydroxide to prepare various secondary amines efficiently. A cesium base not only promoted monoalkylations of primary amines but also suppressed overalkylations. Various amines and alkyl bromides were examined, and the preliminary results demonstrated this methodology was highly chemoselective, favoring mono-N-alkylation over dialkylation. In particular, use of amino acid derivatives afforded the desired secondary amines exclusively.
Among the cesium bases examined, cesium hydroxide monohydrate, in general, gave the highest yields and selectivities although cesium carbonate also worked well, depending on substrates. To confirm the possible cesium effect, defined by the enhanced reactivities in the presence of cesium salts, comparative studies between cesium bases and other bases were performed, demonstrating that cesium hydroxide was superior to other alkali bases tried with the regard to the observed chemoselectivities. Other alkali hydroxides produced moderate yields of the desired product along with a considerable amount of the tertiary amine, whereas cesium hydroxide allowed for a greater selectivity of 9:1 in preference for the monoalkylation product. It was apparent that an unprecedented "cesium effect" in N-alkylation was observed as seen in O-alkylation. Interestingly, the next comparative study implied that the cesium base not only promoted N-alkylation of primary amines, but also inhibited the formation of tertiary amines. According to our experiments, the intended alkylation of a secondary amine (N-butyl-phenethylamine) was very sluggish under our conditions, affording the tertiary amine (N,N-dibutyl-phenethylamine) in only 10% yield, whereas 72% of N,N-dibutyl-phenethylamine was obtained along with the recovery of 25% of N-butyl-phenethylamine in absence of cesium hydroxide after the same duration. Under our cesium base promoted N-alkylation conditions, primary and secondary amines exhibited opposing reactivities, suggesting that the improvement of chemoselectivity would stem mainly from the retarded overalkylation or reversal of normally observed alkylation rates.
Representative Experimental Procedure
To activated powdered 4 Ĺ molecular sieves (500 mg) in anhydrous N,N-dimethylformamide (8.3 mL), was added cesium hydroxide monohydrate (280 mg, 1.7 mmol), and then the white suspension was vigorously stirred for 10 min. After phenethylamine (0.21 mL, 1.7 mmol) was added and followed by additional 30 min of stirring, 1-bromobutane (0.21 mL, 2.0 mmol) was added into the white suspension. The reaction was stirred for 20 h, filtered to remove the molecular sieves and undissolved inorganic salts, and rinsed several times with EtOAc. After the filtrate was concentrated to a nominal volume by blowing air, the residue was taken up in 1 N NaOH, and extracted with EtOAc (4 x 20 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. Flash column chromatography (EtOAc-EtOH, 9:1 v/v) afforded the secondary amine, N-butyl phenethylamine (260 mg, 1.5 mmol; 89%) as a colorless oil as well as tertiary amine, N,N-dibutyl phenethylamine (40 mg, 0.17 mmol; 10%) as a pale yellow oil.