Abstract
Starting from D-phenylalanine, dextroamphetamine sulfate and methamphetamine hydrochloride were synthesized. The reaction sequence proceeds through three intermediates, in which the absolute configuration of the asymmetric carbon atom is changed but the relative configuration remains the same. Either product can be obtained from a common intermediate by altering the reductive conditions employed for the removal of a carbamate protecting group. Following initial observations on the sympathomimetic properties of amphetamine and related compounds1-4, the racemate of amphetamine was introduced into clinical medicine for the relief of nasal congestion5. The (S)-(+)-isomers of amphetamine [dextroamphetamine (V)] and methamphetamine (VI) (Scheme I) have been used in the therapy of obesity, narcolepsy, parkinsonism, and certain behavioral disorders6. Considerable interest also has been generated in the central stimulant properties of these compounds7.
dl-α-Methylphenethylamine and D-α,N-dimethyl-phenethylamine have been prepared by several routes4,8-12. The deuterium-labeled analogs of these compounds also have been synthesized13,14. Karrer and Ehrhardt15 originally prepared V from D-phenylalanine via the N,O-di-p-toluenesulfonate derivative of II. Using a modification of that procedure, Gal16 recently synthesized the deuterium analog of V and the corresponding (R)-(-)-isomer for use as internal standards in GLC-mass spectral studies. An asymmetric synthesis of V also was described17.
An earlier report18 presented the synthesis of (R)-(-)-and (S)-(+)-α-methyltryptamine from the corresponding tryptophan isomers. The present work outlines the application of this method to another aromatic alpha-amino acid, phenylalanine.
The reaction sequence (Scheme I) is essentially the same as that used previously18. In contrast to the corresponding indole analog, the N-benzyloxycarbonyl-O-p-toluenesulfonyl intermediate (IV) is a stable, crystalline solid. The substitution of nitrogen with a carbamate group has the advantage of providing either the primary (V) or secondary (VI) amine, depending on the conditions employed for the reduction of IV. Although the preparation of N-methylamino acids by lithium aluminum hydride reduction of their N-benzyloxycarbonyl derivatives has been known for years19, this reaction has seldom been applied to other types of amines20.
Experimental
(R)-(+)-2-Amino-3-phenylpropanol (II)
To a suspension of 1.3g (34 mmoles) of lithium aluminum hydride in 75 ml of anhydrous tetrahydrofuran was added, in portions, 2.1 g (12.7 mmoles) of D-phenylalanine. After the addition, the reaction mixture was refluxed for 20 min and cooled to room temperature. Then the complex and excess reagent were decomposed by dropwise addition of 2N aqueous sodium hydroxide and water. The white solids were collected by filtration and washed with 100 ml of tetrahydrofuran.

The filtrate and washings were combined and concentrated under reduced pressure. The residual clear oil slowly crystallized and was recrystallized from ethyl acetate-hexane, 1.75 g (91%), mp 90-91°C [lit.21 mp 91-92°], [α]D 23.8° (c 1.0, ethanol) [lit.19 [α]D 24.6°].
(R)-(+)-N-(Benzyloxycarbonyl)-2-amino-3-phenylpropanol (III)
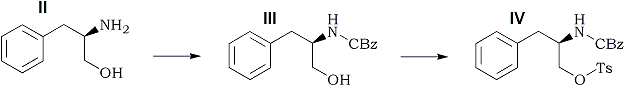
A mixture, of 1.5 g (9.9 mmoles) of II and 1.12 g (10.6 mmoles) of sodium carbonate in 25 ml of acetone and 25 ml of water was stirred at room temperature, and 1.5 ml (10.5 mmoles) of benzyl chloroformate was added. The reaction mixture was stirred for 3.0 hr, diluted with 50 ml of water, and acidified (to pH 2) with concentrated hydrochloric acid. The mixture was shaken with 300 ml of ethyl acetate, and the organic phase was washed with 100 ml of saturated aqueous sodium chloride. After drying (magnesium sulfate), the filtered organic solution was concentrated in vacuo. The product was crystallized from ethyl acetate-hexane, 1.5 g (55%), mp 91-92°C, [α]D 41.3° (c 1.0, ethanol).
(R)-(+)-N-(Benzyloxycarbonyl)-2-amino-3-phenylpropanol p-Toluenesulfonate (IV)
Compound III (1.25 g, 4.4 mmoles) and p-toluenesulfonyl chloride (955 mg, 5 mmoles) were dissolved in 100 ml of pyridine The solution was stored at room temperature with the exclusion of moisture for 4 days. Water (2.0 ml) was added; after 30 min, the solvent was distilled under reduced pressure. The residue was partitioned between 300 ml of ethyl acetate and 75 ml of saturated aqueous sodium bicarbonate, and the organic phase was dried (magnesium sulfate). After filtration of the drying agent and concentration of the filtrate, the residual semisolid was preparatively chromatographed on two 1-m x 20-cm glass plates coat with 750-µm layers of silica gel GF, using 5% acetone in benzene as the developing solvent. The product band was removed from the plates and eluted with ethyl acetate. Concentration of the eluate afforded a clear syrup, which was crystallized from ethyl acetate-hexane, 985 mg (50%), mp 96-97°C, [α]D 29.7° (c 1.0, ethanol).
Dextroamphetamine [(S)-(+)-α-Methylphenethylamine] (V) Sulfate
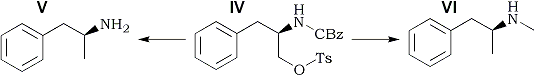
Compound IV (250 mg, 0.568 mmole) and 100 mg of 10% Pd/C were mixed in 30 ml of absolute ethanol, and the reaction was shaken under 50 psi of hydrogen for 1.0 hr. The catalyst was removed by filtration (celite), and the filtrate was concentrated in vacuo. The residue was partitioned between 30 ml of 1N aqueous sodium hydroxide and 200 ml of ethyl acetate. The organic solution was washed with 50ml of water.
The dried (magnesium sulfate) solution was concentrated under reduced pressure (bath temperature of 25°C), and the oily residue was distilled in vacuo (Kugelrohr apparatus) at 0.05 mm and 40-60°. The clear distillate was dissolved in 3.0 ml of ether and carefully acidified (to pH 4) by addition of 0.2N H2SO4 in ethanol. The white solid was collected by filtration, washed with ether, and dried in vacuo to give 40 mg (38%), mp >300°, [α]D 20.1° (c 1.0, water) [lit.22 [α]D 21.5°]. The IR spectrum was identical to that reported for this compound23.
Methamphetamine [(S)-(+)-α,N-Dimethylphenethylamine] (VI) Hydrochloride.
To a stirred suspension of 200 mg (5.26 mmol of lithium aluminum hydride in 5.0 ml of tetrahydrofuran was added a solution of 250 mg (0.568 mmole) of IV in 5.0 ml of tetrahydrofuran, the reaction mixture was refluxed for 30 min and cooled to room temperature and the complex was decomposed by careful addition of 2 N aqueous sodium hydroxide and water. After filtration of the white solids the filtrate was concentrated in vacuo. The residual oil was distilled in vacuo (Kugelrohr apparatus) at (0.05 mmHg and 40-60°C. The distillate was dissolved in 2.0 ml of ether, and the solution was acidified by addition of saturated hydrogen chloride in ether. The crystalline plates were collected by filtration, washed with ether, and dried in vacuo, yielding 65 mg (62%), mp 171-173°C [lit.9 mp 172°C, [α]D 15.1° (c 1.0, water) [lit.24 [α]D 14-20°]. The IR spectrum was the same as that reported for this compound23.