In connection with our synthesis of analogs of serotonin neurotoxins1 we needed access to 4-hydroxy-3-methoxy-5-methylbenzaldehyde (1) in quantities. The only previous method2 for the synthesis of 1 involved conversion of o-vanillin to 2-hydroxy-3-methoxytoluene followed by formylation using CHCl3/NaOH in 12% overall yield. We thought a more efficient process would result if we could introduce a methyl group selectively in the position ortho to the hydroxyl group of vanillin (2). However, no direct and selective method for introducing a methyl group into a benzene ring containing a hydroxyl as well as an aldehyde group has been described in the literature. In this paper we describe the development of a method for the synthesis of 1 from vanillin (2) without requiring the protection of either the hydroxyl or the aldehyde function and demonstrate the generality of such a method.
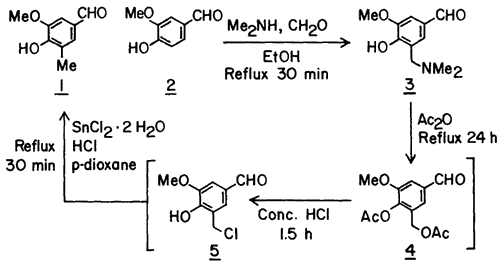
In devising the present method we took advantage of the selectivity and the high yield that can often be realized in the Mannich reaction of phenols3. Thus in the first step in the synthesis of 1 from 2, the methyl group equivalent, dimethylaminomethyl group, was introduced in vanillin in 86% yield to give crystalline Mannich base 3. Previous methods for the synthesis of C-methylated phenols from phenolic Mannich bases have involved catalytic hydrogenolysis of the Mannich base itself4, its hydrochloride salt5 or its acetolysis product6 (cf. 4) and NaBH3CN reduction of the methiodide of the Mannich base7. None of these procedures, due to their incompatibility with the aldehyde function, could be applied to the conversion of 3 to 1.
We next considered the conversion of the dimethylaminomethyl group of 3 to a methyl group equivalent that can be reduced to a methyl group in the presence of a free aldehyde function. The chloromethyl group appeared to be such a group based on Sandin and Fieser's8 observation that the iodomethyl group of 9-methyl-10-iodomethyl-1,2-benzanthracene can be converted to a methyl group using SnCl2 in conc. HCl.
For the synthesis of the chloromethyl derivative9 5. Mannich base 3 was first treated with excess acetic anhydride under reflux to give the diacetate 4. Without its isolation, but after the removal of excess acetic anhydride, the diacetate 4 was treated with conc. HCl at ambient temperature for 1.5 h to generate the chloromethyl derivative 5 which precipitated out of the reaction mixture. 1H-NMR spectrum of the crude precipitate suggested it to be 5, however, due to its instability, an analytical sample could not be prepared. The chloromethyl derivative, in turn, without its isolation was reacted with SnCl2·2 H2O in a mixture of conc. HCl and p-dioxane under reflux for 30 min to give, after sublimation of the crude product, the desired C-methylated product 1 in 85% yield based on the Mannich base.
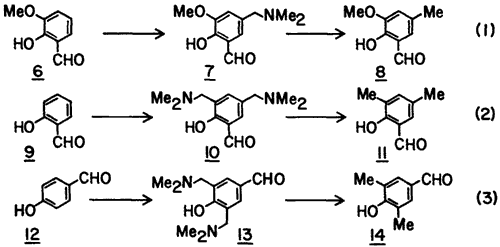
To determine the generality of the procedure, we studied three other examples (Eq. 1-3). In each case the Mannich base was converted to the corresponding C-methylated derivative without the isolation of the intermediate acetolysis product (cf. 4) or the chloromethyl derivative (cf. 5). These examples show that the method is suitable for the introduction of the methyl group para to the hydroxyl group (Eq. 1-2) and also suitable for bis-C-methylation (Eq. 2-3).
Experimental
5-Dimethylaminomethyl-4-hydroxy-3-methoxybenzaldehyde (2)
To a stirred solution of 37% aqueous CH2O (120 g, 1.5 mol) and 40% aqueous Me2NH (180 g, 1.5 mol) in 900 mL of EtOH was added vanillin (2, 152 g, 1 mol). The mixture was refluxed for 30 min, stirred at 25°C for 24 h and then refrigerated overnight. The white granular solid was collected by filtration, washed with ice-cold acetone and then dried under vacuum to give 179.8 g (86%) of 3: mp 139-141°C.
4-Hydroxy-3-methoxy-5-methylbenzaldehyde (1)
A solution of the Mannich base 3 (104.5 g, 500 mmol), in 500 mL of Ac2O was refluxed for 24 h protected from moisture. Volatile materials were removed by distillation (bp 55-80°C) under aspirator vacuum. After cooling the residue (which was the crude diacetate 4) to ~40°C, 500 mL of conc. HCl was added gradually and then the mixture was stirred at ambient temperature for 1.5 h by which time most of the chloromethyl derivative 5 had precipitated. Enough p-dioxane (~400 mL) was added to ensure dissolution of the solid at 60-70°C. Then with efficient (mechanical) stirring 337.5 g (1.5 mol) of SnCl2·2 H2O was added and the mixture was refluxed for 30 min. After cooling to 25°C and dilution with 200 mL conc. HCl, the mixture was extracted with CHCl3 (5 x 150 mL). The combined organic layers were washed with 6 N HCl, water, 10% NaCl solution and then evaporated in vacuo to dryness. The crude residue was adsorbed on silica gel (63-200 mesh, 70 g) by adding silica gel to a solution of the crude in CHCl3 and then evaporating the solvent in vacuo. A slurry of the mixture in CHCl3 was then applied on a column of silica gel (100 g) and eluted with ether-hexane (2:1). Evaporation of eluents followed by sublimation (0.1 mm, 120°C) of the grey solid residue gave 70.6 g (85%) of 1 as a white granular solid: mp 99-101°C (lit.2 mp 99-100°C).
5-Dimethylaminomethyl-2-hydroxy-3-methoxybenzaldehyde (7) was prepared from o-vanillin (6, 3 g) as described for 3 except that reflux was carried out for 4 h and then the mixture was evaporated in vacuo to dryness. The residue was triturated with Et2O whereby a light yellow solid was formed which was thoroughly washed with Et2O and dried (yield 78%): mp 110-112°C; mp of HCl salt 170-172°C (lit.11 mp 169-172°C).
2-Hydroxy-3-methoxy-5-methylbenzaldehyde (8) was produced from 7 (2.8 g) in 63% yield: mp 74-76°C (lit.12 mp 76°C).
3,5-Bis(dimethylaminomethyl)-2-hydroxybenzaldehyde (10) was prepared from 9 (4 g) as described for 7 except that three equivalents each of CH2O and Me2NH were used and was isolated as an oil (87%).
3,5-Dimethyl-2-hydroxybenzaldehyde (11) was produced from 10 (3.8 g) using six equivalents of SnCl2·2H2O at the reduction step, in 55% yield: mp 12-14°C (lit.13 mp 12-14°C).
3,5-Bis(dimethylaminomethyl)-4-hydroxybenzaldehyde (13) was prepared from 12 (4 g), as described above for 10, as a syrup in 83% yield.
3,5-Dimethyl-4-hydroxybenzaldehyde (14) was prepared from 13 (3.8 g) as described above for 11 in 63% yield: mp 113-115°C (lit.14 mp 114-115°C).