Abstract
Synthesis of 3,4-dimethoxy-5-hydroxybenzaldehyde (1) in three steps from vanillin with the key step being a copper catalyzed hydrolysis of 5-bromovanillin to give 4,5-dihydroxy-3-methoxybenzaldehyde.
A series of novel hydroxybutenolide compounds with activity as endothelin antagonists was discovered recently in our endothelin program. These compounds are non-peptides and have selective activity at nanomolar concentrations with Endothelin A receptors.1 We required a method to prepare 3,4-dimethoxy-5-hydroxybenzaldehyde (1) as an intermediate to the hydroxybutenolides as part of this program. The synthesis of 1 has also been of considerable interest to groups working in other areas such as gallolignins and flavonoids.2,3,4 In this paper we will describe the development of a short and facile procedure that allows preparation of the desired benzaldehyde on moderate scale.
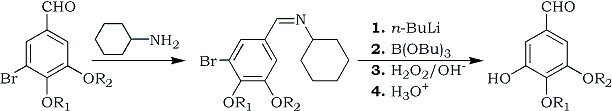
In our initial work, small quantities of 1 were prepared using the method of Jacob and Shulgin2a which is shown in Scheme 1. Their synthetic route involved 6 steps starting from vanillin (2) with an overall yield of 51%. This route involved protecting the aldehyde group as its cyclohexylimine (3), which was purified by vacuum distillation. The purified imine was then converted to 1 in four steps using a one-pot sequence. The protected bromovanillin, 3, was lithiated with butyllithium and immediately treated with tributyl borate. After warming to room temperature, the borate intermediate was reacted with 30% hydrogen peroxide and then quenched into dilute acid to give 1. Evaluation of this method identified several potential problems for scale-up. The imine 3 is very high boiling requiring very good vacuum (<1 mmHg). A low temperature system is required for the lithiation and boronation, which are done at -78°C. Finally, the peroxide reaction is quite exothermic and probably difficult to handle on large scale. With these considerations in mind we started looking for alternate routes to 1.
{kind=link}
Several other synthetic routes to 1 have been published. Synthetic routes to 1 from gallic acid derivatives in 5 steps in less than 25% overall yield were described Mauthner3a and also by Battersby.3b Both routes required Rosenmund reduction of benzoyl chloride intermediates to aldehydes to complete the syntheses. The low overall yields of these routes made them unattractive. More recently, Iinuma described a route to 1 from o-vanillin (4) that involved 5 steps in an overall yield of approximately 20% shown in Scheme 2.4 The key reactions in this route are a Baeyer-Villiger reaction with peroxyformic acid followed by a Rosenmund-von Braun cyanation with cuprous cyanide run at 200°C. There was no advantage to this route either since it had a low overall yield combined with the use of column chromatography and dangerous reagents such as peroxyformic acid and cuprous cyanide to prepare 1.
We wanted a scalable route with a high degree of atom efficiency. In planning this synthesis we also hoped to avoid undesirable chromatography, high vacuum distillations and temperatures outside a range of -40°C to 150°C, all of which are used in the literature methods.
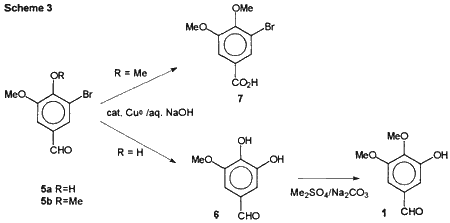
We have now developed a three step synthesis of 1 from vanillin shown in Scheme 3 which achieved these goals. The first intermediate was 5-bromovanillin (5a) which is commercially available or can be prepared by bromination of vanillin with bromine in acetic acid by the method of Dakin.5
In the key reaction, 5a was converted to 3-methoxy-4,5-dihydroxy- benzaldehyde (6) using copper catalyzed base hydrolysis. The hydrolysis of 5-bromovanillin has been done by several groups by heating the reaction mixture containing stoichiometric quantities of copper powder in an autoclave at 210°C for 1 or 2 hours with isolated yields of approximately 40%.6 We found that the reaction can be done under milder reaction conditions than previously reported. To accomplish the hydrolysis, 5a is heated in 8% sodium hydroxide solution with 1.5 mole% copper powder at 101°C for 24-27 hours, using a modification of the method of Wehrli.7 The desired 6 was isolated using continuous extraction with ethyl acetate and obtained in 60-70% yield after recrystallization from toluene. This hydrolysis required the presence of the 4-hydroxy group to give the desired hydrolysis. When the same reaction conditions were used with the 4-methoxy analogue (5-bromo-veratraldehyde, 5b), the aldehyde group reacted in a Cannizzaro reaction to give 5-bromo-3,4-dimethoxybenzoic acid (7) in good yield as previously reported by Schriner and McCutchan.8
Methylation of 6 with 1.1 equivalent of dimethyl sulfate and sodium carbonate in acetone gave the desired benzaldehyde in 70% yield. Only trace amounts of the regioisomeric product, syringaldehyde (3,5-dimethoxy-4-hydroxybenzaldehyde), were noted by TLC. The primary by-product is 6-10% of the over-methylated product, 3,4,5-trimethoxybenzaldehyde, from which the desired product was easily removed by alkaline extraction followed by acidification. The crystalline product can be recrystallized from either toluene-heptane or methanol-water.
In conclusion, we have developed a convenient synthesis of the useful synthetic intermediate 3,4-dimethoxy-5-hydroxybenzaldehyde that can be run on moderate scale and is operationally scalable. Our overall yield is comparable to the method of Jacob and Shulgin but our method avoids undesirable distillations and low reaction temperatures. In addition, our method is safer because it minimizes the use of toxic or hazardous reagents. The one dangerous reagent common to all the routes is dimethyl sulfate. This reagent is essentially unavoidable since all the routes require preparation of methyl ethers. However, it is the least volatile of the typical methylating agents and thus the easiest to control on large scale.
Experimental
All of the reagents used in this study were purchased from the Aldrich Chemical Company. All of the reactions were run under a nitrogen atmosphere. The 1H spectra were taken on a Varian 200 MHZ spectrometer, chemical shifts are expressed in ppm from tetramethylsilane as an internal standard. The melting points were determined on a Buchi melting point apparatus and are uncorrected.
3-Methoxy-4,5-dihydroxybenzaldehyde (6)
5-bromovanillin (5) (200 g, 0.91 mol), sodium hydroxide (245 g, 6.1 mol) and copper powder (1 g, 0.016 mol) were slurried into 3 L water. The reaction mixture was heated at reflux for 24-27 hours. Sodium hydrogen phosphate (4.5 g, 0.032 mol) is added for the last half hour of reflux. The reaction is then cooled to less than 50°C, filtered to remove a precipitate of cupric hydrogen phosphate and acidified with hydrochloric acid (460 g). The reaction mixture was placed in a continuous extractor and extracted with ethyl acetate (3 L). The ethyl acetate extract was stirred with activated carbon and filtered. The filtrate was washed with saturated aqueous EDTA solution followed by salt. The solution was then dried over magnesium sulfate and filtered. The ethyl acetate solution was concentrated to a crude solid. The crude product was dissolved in boiling toluene (2 L), treated with activated carbon, filtered and cooled to crystallize. The product, 3-methoxy-4,5-dihydroxybenzaldehyde, was isolated in approximately 60% yield (86 g) with a mp 132-133°C (lit.7a mp 132-134°C); 1H NMR (200 MHz, d6-DMSO) 3.7 (3H, s, OCH3), 6.98 (1H, d, ArH, J=9Hz), 7.01 (1H, d, ArH, J=9Hz), 9.5 (2H, s, ArOH), 9.6 (1H, s, CHO).
3,4-dimethoxy-5-hydroxybenzaldehyde (1)
3-Methoxy-4,5-dihydroxybenzaldehyde (100 g, 0.595 mol), dimethyl sulfate (75.0 g, 0.595 mol) and sodium carbonate (69.4 g, 0.654 mol) were slurried into acetone. The reaction mixture was heated at reflux for 4-6 hours and then cooled to room temperature. After filtering to remove inorganic salts, the acetone was removed by distillation and replaced with toluene. The toluene solution was stirred at room temperature for one hour and filtered to remove an insoluble tar. The product was extracted with a dilute sodium hydroxide solution. The water layer was separated, acidified with hydrochloric acid to a pH of 1-2 and extracted with toluene. The toluene layer was dried with magnesium sulfate then treated with activated carbon and filtered. The toluene solution was concentrated to an oil which readily crystallizes to give 1 in 70% yield (75.8 g). The product can be recrystallized, if necessary, from a 2:1 mixture of toluene/heptane. 3,4-dimethoxy-5-hydroxybenzaldehyde was obtained with mp 65.6-66.5°C (lit.4a mp 64-65°C); 1H-NMR (200 MHz, CDCl3) 3.91 (3H, s, OCH3), 3.98 (3H, s, OCH3), 6.1 (1H, S, ArOH), 7.0 (1H, d, ArH, J=9Hz), 7.1 (1H, d, ArH, J=9Hz), 9.8 (1H, d, CHO).