Abstract
With p-cresol as starting material, 3,4,5-trimethoxybenzaldehyde was synthesized in an overall yield of 67.4% via bromination, hydrolysis, methoxylation and methylation.
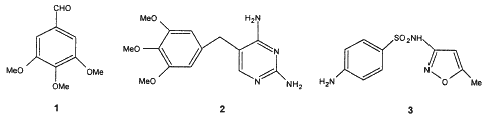
The most important use of 3,4,5-trimethoxybenzaldehyde 1 as a pharmaceutical intermediate is to prepare the antibacterial agent trimethoprim 2,1-2 which is a potent and selective inhibitor of bacterial dihydrofolate reductase. Trimethoprim is used solely, or in combination with sulfamethoxazole 3, to treat a wide range of bacterial infections in humans and poultry.
Previous synthesis of 1 has been achieved simply from vanillin by bromination, followed by a cuprous chloride-catalyzed displacement of bromine by methoxide.3 Another synthesis of 1 has been developed through the liquid-phase oxidation of 3,4,5-trimethoxytoluene to the corresponding aldehyde with Co(OAc)2–Mn(OAc)2 as catalyst.4 Although 1 can be obtained in high yields and fewer steps, the relatively high cost of starting material vanillin and 3,4,5-trimethoxytoluene necessitates further work to find an efficient and convenient approach to 1 for a practical industrial process. Manchand et al.5 reported an attractive method of the preparation of 1 that relies on the methylation of syringaldehyde 7, which was synthesized from p-cresol via a series of reactions shown in Scheme 1.
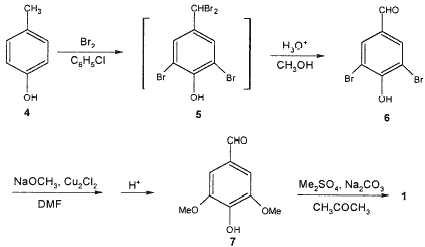
Scheme 1.
However, low yield (60%) of 3,5-dibromo-4-hydroxybenzaldehyde 6, large amount consumption of solvent DMF due to its liability to thermolysis in the presence of CH3ONa during the methoxylation of 6, the use of the solvent acetone and long reaction time (18 h) in the methylation of 7 restrict its application as an efficient industrial process. Therefore, a more economical, convenient and environment-friendly synthesis of 1 is needed.
In general, the requirement of solvents such as hexamethylphosphorous triamide (HMPT), DMF and pyridines and the requirement of copper-catalyst characterize the methoxylation of non-activated aromatic bromides.6-7 It is difficult to increase reflux temperature due to the release of “solvent cage bonded” methanol from sodium methoxide.8 The low reflux temperature and the low CH3ONa concentration disadvantage the progress of the methoxylation.
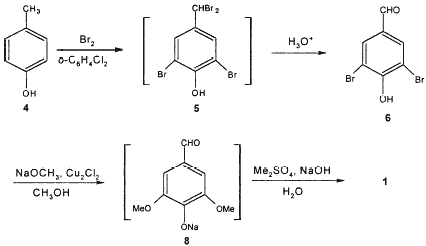
Scheme 2.
We improved herein a procedure of preparation of 1.
As Scheme 2 shows, the bromination of 4 in the solvent o-dichlorobenzene at 40°C and then at 160°C respectively gave the tetrabromide 5, and then the direct hydrolysis of 5 with H2O afforded 6 in an overall yield of 81.6% from 4. Using o-dichlorobenzene instead of chlorobenzene as solvent and one-pot method for the synthesis of 6 were one character of our procedure. GC/MS analysis shows that major by-product formed in the synthesis of 6 was 3,5-dibromo-4-hydroxybenzoic acid (<1%) while another possible by-product 3,5-dibromo-4-hydroxybenzyl alcohol was not detected. de la Mare and Newman9-10 reported that the hydrolysis of 2,6-dibromo-4-dibromomethylphenol 5 was very rapid to give 6, which was easily oxidized to afford the corresponding acid. It is consistent with our experimental results. Treatment of 6 with 3.7–3.9 equivalents of freshly prepared sodium methoxide (ca. 28–30 wt%) in the presence of catalytic amount of cuprous chloride11 and DMF at 120°C12 in autoclave gave sodium phenolate of 3,5-dimethoxy-4-hydroxybenzaldehyde 8. The sodium phenolate was directly methylated with dimethylsulfate in NaOH aqueous solution, affording 1 in the yields of 80.0–82.6%. Methanol was used as solvent for methoxylation of 6. Only small amount of DMF was used as a co-solvent. Using H2O as solvent for the methylation of 8, NaOH can act as a good acid-capturing agent and thereby the reaction time was shortened to 2–3 h. Hence, conventionally used solvent acetone is unnecessary. The use of the high concentration of sodium methoxide and the utilization of the adequate crystallizability of 8 for isolation are the other two characters of our procedure.
Experimental
Melting points were determined in capillaries on a domestic melting point apparatus and are uncorrected. Silica gel plates were used for analytical TLC and the spots were visible with iodine vapor.
3,4-Dibromo-4-hydroxybenzaldehyde 6
3-Necked round-bottomed flask equipped with a condenser attached to a calcium chloride drying tower, a mechanical stirrer, a thermometer, and a dropping funnel was charged with a solution of 43.2 g (0.40 mol) of p-cresol in 200 mL of o-dichlorobenzene. The solution was cooled to 10°C and treated during 1 h with a solution of 130.0 g (0.81 mol) of bromine in 100 mL of o-dichlorobenzene at such a state that the temperature was kept below 40°C. The mixture was stirred, heated at 160°C and simultaneously dropped with another solution of 130.0 g (0.81 mol) of bromine in 100 mL of o-dichlorobenzene during 3 h. This deep red mixture was sequentially stirred for 30 min. After being cooled to room temperature and 145 mL of water being added, the mixture was heated to 120°C and kept at the temperature for 4 h. After being cooled to 5°C, the mixture was separated into upper organic layer, middle aqueous layer and lower solid layer. The solid product was collected by filtration and washed with water up to neutrality. It was dried in vacuo at 50°C to give 91.4 g (81.6%) of the off-white solid 6, mp 180–182°C (lit.5 179–182°C).
3,4,5-Trimethoxybenzaldehyde 1
About 142.0 g (0.74–0.79 mol) of freshly prepared 28–30 wt% sodium methoxide, 8 mL of DMF, 4 g of cuprous chloride and 56.0 g (0.20 mol) of 6 were added into a 500 mL magnetically stirred stainless steel autoclave. After the air was replaced with N2, the autoclave was heated to 120°C and kept at the temperature for 3 h. After being cooled to room temperature, the reaction mixture was taken out from the autoclave and transferred into a round-bottomed flask. Solvents were evaporated from the flask in vacuo below 60°C, and the residue was treated with 200 mL of water. The mixture was stirred and heated to 90°C for dissolution, then cooled to 0°C for crystallization of 8. After NaBr dissolved in the stock was removed by filtration, yellow green sodium phenolate 8 was obtained. The filter cake was dissolved in 300 mL of water, added with 1.5 g of active carbon, and then heated to 95°C for 30 min. After hot-filtration for removing the filter residue (catalyst included), the filtrated stock was cooled to 40°C, and was treated by slowly dropping 50.0 g (0.40 mol) of dimethylsulfate and 30 wt% 112 g (0.84 mol) of NaOH aqueous solution during 2 h. The pH value should be carefully controlled within 8.5–9.3. Stirring was continued at 50°C for 30 min and then at 0°C for 30 min, and the product was collected by filtration. It was washed with water up to neutrality and dried in vacuo at 45°C to give 32.4 g (82.6%) of 1 as colorless crystals, m.p. 74–75°C (lit.5 73–75°C).