2C-B synthesis without LAHby BeakerIntroductionThe following is a synthesis of 2C-B from 2,5-dimethoxybenzaldehyde that does not require the use of the slightly hazardous and/or difficult to obtain reagents normally associated with its synthesis, notably LAH, pressurized H2, and Br2. No doubt some clandestine chemists have been discouraged from attempting the synthesis of what is, IMHO, a pretty cool substance by the nature of these reagents and the lack of a clearly written procedure for an alternate route that does not use them. However, alternate routes do exist, and one of them is detailed below. Note that this route does require one additional step to acheive the nitrostyrene reduction than with the use of H2 or LAH, but that the yield is actually substantially higher than what others have reported with those reducing systems. Also, the bromination procedure is somewhat unrefined at present and does not result in the greatest of yields or the easiest of workups, so feel free to use the classical procedure if you want to make or buy your own bromine. As a final note, although this route will happily accomodate batch sizes of ~50g in 2L glassware, it does not scale anywhere near as well as catalytic hydrogenation, so if you're trying to go huge, it's probably not for you. ProcedureStep 1: Condensation of 2,5-dimethoxybenzaldehyde with nitromethane 
In a 500mL RBF equipped with a reflux condenser and a stir bar, place 100g 2,5-dimethoxybenzaldehyde, 15g ammonium acetate, and 250mL of nitromethane. Heat to a gentle reflux while magnetically stirring. Maintain reflux for ~45min, by which time the color of the solution should progress from clear/yellow to a deep reddish-black. Remove heat and carefully pour the hot rxn mixture into 1L of ice-cold 70% IPA. Allow the IPA/rxn mixture to stand for a while. You should now have a flask full of orange solids floating in red/black mother liquor. Vacuum-filter the solids and wash them with additional portions of ice-cold 70% IPA until the filtrate is no longer reddish. Thoroughly dry the collected orange solids by pulling air through the filter for a while and then under vaccum. It is very important that the nitrostyrene be completely dry before proceding to the next step. Yield - 106.1g (84%) of 2,5-dimethoxynitrostyrene Purity - Single spot by TLC, NMR is clean Step 2: Sodium borohydride reduction of 2,5-dimethoxynitrostyrene 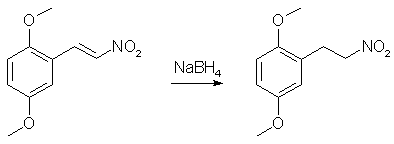
Into a dry 2L RBF flask equipped with a stir bar was added 400mL of anhydrous ethanol (If you can't get anhydrous ethanol, use anhydrous IPA (Do not use methanol!!!). The rxn was cooled to 0°C in an ice/water bath and 36.2g of sodium borohydride was added (slight H2 evolution). A pressure-equalized addition funnel was charged with a pre-made saturated solution of 50g 2,5-dimethoxynitrostyrene in THF (about 600mL) and attached to the flask. A piece of tubing was attached to the top of the addition funnel and run outside to vent the hydrogen that is will evolve during the course of the reaction. While maintaining the ice/water bath, slowly (reaction is exothermic, so go slowly) all of the bright yellow nitrostyrene solution (refill the addition funnel if necessary) was added to the sodium borohydride solution over the course of ~90 min (Note: gas will evolve over the course of the addition. It is H2. Be careful). After the addition is complete, the rxn was allowed to stir for an additional 10 min and then poured into a 4L erlenmeyer containing 1L of H2O and a 3" stir bar (H2 evolution). While stirring, 250mL glacial acetic acid (Heavy H2 evolution) was carefully added (one could use 400 mL 31.45% HCl). The quenched reaction mixture was divided into three portions. In a 2L sep funnel, each portion was combined with 500mL Et2O (or toluene) and 500mL brine. The funnel was shaken and the aqueous (bottom) layer was discarded. The organics were washed with 3 additional 500mL portions of brine. This was repeated with the other two portions. The organics were combined, dried over MgSO4, filtered and the solvent evaporated to give a clear yellow oil. Yield - 47.0g of crude 2,5-dimethoxynitroethane Purity - Two spots by TLC. NMR analysis indicates a 50:1 molar ratio of the desired product to dimeric impurity (this is the only impurity present). Adjusted yield of 2,5-dimethoxynitroethane is 45.2g (89.5%). Step 3: Catalytic Transfer Hydrogenation of Crude 2,5-dimethoxynitroethane 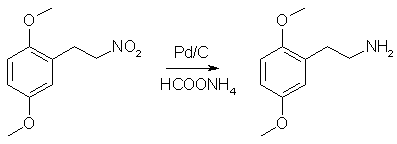
The crude product of the previous step was dissolved in 400mL MeOH and placed in a 1L RBF equipped with a stir bar. In a separate beaker away from all combustible materials, 1g of 10% Pd/C was carefully wetted down with MeOH and the resulting slurry transferred to the rxn flask. To the rxn flask was added 62g ammonium formate. The flask was equipped with a reflux condensor, a piece of tubing was attached to the top of the condensor, and the end of the tubing was submenged in a container of water (this works to exclude O2 from the rxn while allowing the evolving CO2 to escape). The rxn was gently refluxed for 24 h (CO2 evolution), cooled, filtered through celite to remove the Pd/C, and the solvent evaporated. The residue was taken up in 150 mL of Et2O (or toluene) and 300 mL of H2O and the pH adjusted to >12 with 20% NaOH. The mixture was transfered to a sep funnel, shaken, and separated. The aqueous layer was extracted with 2x100 mL portions of Et2O (or toluene). The combined organics were dried over MgSO4, filtered, and gassed with HCl (2C-H*HCl is partially soluble in DCM, so don't gas in that solvent). The resulting white crystalline solids were filtered, washed with Et2O, and allowed to air dry to give 2C-H Hydrochloride. Yield - 43.8g (94%) of 2C-H Hydrochloride Purity - Single spot by TLC. NMR is clean. Step 4: Bromination of 2C-H Freebase 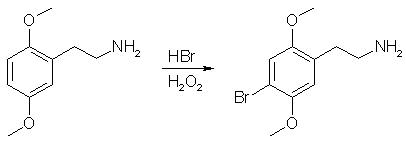
The 2C-H*HCl was dissolved in a 300 mL H20. The pH was adjusted to >12 with 20% NaOH and the aqueous layer was extracted with 4x100 mL DCM. The DCM was evaporated to give 2C-H freebase, which was dissolved in 500 mL of 3:1 AcOH/H2O. The rxn was cooled to 0C in an ice/water bath. 37.3g of 48% aq. HBr was added, followed immediately by 23.8g of 30% H2O2. The rxn was stirred for 6 hr, allowing the ice bath to melt. The majority of the AcOH was removed under vacuum and the nasty reddish-black rxn mixture was partitioned between 1L H20 and 500 mL EtOAc (EtOAc was found to be much better for dissolving the impurities in this rxn than Et2O or toluene. This is messy at first, but everything should go into solution after much agitation). The layers were separated and the aqueous extracted with an additional 500 mL EtOAc. The aqueous was basified to pH >12 with 20% NaOH and extracted with 3x200 mL portions of Et2O. The combined organics were washed with 400 mL brine, dried over MgSO4, filtered and gassed with HCl. The resulting tan crystalline solids were filtered and recrystalized from boiling 1:1 IPA/Toluene to give pure 2C-B*HCl as a white crystaline solid. Yield - 34.0g (57%) of 2C-B Hydrochloride Purity - Single spot by TLC. NMR is clean. Before some jackass reads this procedure and asks me why 4 equivalents of sodium borohydride is used for the reduction, here is a interesting little table for your reading pleasure.
Sunlight's Voice: After several failures, we finally got an interesting success using Beaker's 2C-H synth. We post it due me and others had problems with it. The rxn was carried out exactly as Beaker posted, no problems, everything goes fine. We made the sulfate salt instead of the hydrochloride by adding a 1:10 H2SO4:IPA v/v to the toluene extracts until the pH was slightly acidic. Yield was 65% from the nitrostyrene, not the >80% of Beaker, but anyway really interesting. By the way, we have never got a 84% of the nitro in step one, only 65-75% and reacting 2.5 hours, not 45 minutes, at 45 minutes the rxn seemed to be incomplete. The reason of the success is the catalyst, all attempts with homemade catalyst gave miniscule yields, so we decided to buy it from Aldrich, 10% Pd/C on activated carbon, and now rxn finally works. In the reduction with Pd/C and ammonium formate, solid crystals are formed in the condenser (we guess they are ammonium carbonate) and they can clog the condenser. We had a little hazard in a previous test, so you need to control it and remove the crystals. We doubled the catalyst in order to finish rxn in 12 hours instead of 24, but rxn finish in about 5 hours, but we let it 12 hours. Other thing, the catalyst was washed with methanol, and when it was drying, it catched fire, next time we'll make a final wash with water before letting it dry, so we could reuse it. Barium's Voice: For better yields: 10g (47,4mmol) 1-(2,5-dimethoxyphenyl)-2-nitroethane was dissolved in 50ml EtOH containing 300mg 5% Pd/C in a 250ml rb flask. 14g (166mmol) potassium formate was dissolved in 9ml water (500mmol, roughly 3 eq) and this solution was added to the alcohol solution in one portion. The mixture was heated to 70-75deg C for 5 hours with good stirring. During this time the mixture became thicker and thicker due to the precipitation of KHCO3. After three hours the mixture was too thick to be properly stirred so another 50ml EtOH was added. When 5 hours had passed the the reaction mixture was cooled to roomtemp and acidified to pH2 with dilute HCL , celite added and the catalyst removed by filtration. 100ml water was added to the filtrate and then it was extracted with 2x100ml toluene. The aqueous phase was the basified to pH 12 with 50% aq. NaOH and extracted with 2x100ml toluene. The organic phase was dried with MgSO4 and removed in a rotovap leaving a yellow oil. This oil was dissolved in 100ml EtOAc and gassed with dry HCL. Yield 6,6g (30,3mmol, 64%) 2,5-dimethoxyphenylethylamine HCl, mp 139-140°C. In the presence of a catalyst hydrogen is formed according to: HCO2K + H2O __> H2 + KHCO3 For further reading check US patent 4,792,625 |