Abstract
By use of appropriate reactions and sequence of steps, 2,5-dimethoxybenzaldehyde can be converted either to 4-(bromo or iodo)-2,5-dimethoxybenzonitrile or 2-(bromo or iodo)-3,6-dimethoxybenzonitrile.
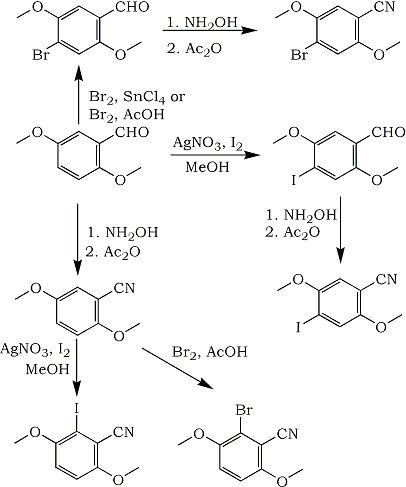
In the course of other work, we needed to prepare either 4-bromo-2,5-dimethoxybenzonitrile or 4-iodo-2,5-dimethoxybenzonitrile. Neither of these compounds had been previously reported in the literature. Two possible pathways to these compounds were pursued, both of which began with 2,5-dimethoxybenzaldehyde (1).
In the first pathway, 1 was brominated to form 4-bromo-2,5-dimethoxybenzaldehyde (2) using either the procedure of Barfknecht and Nichols1, or by treatment with bromine in acetic acid. Neither of these procedures worked in high yield. Treatment of 2 with hydroxylamine hydrochloride and sodium hydroxide in ethanol yielded the oxime, which was converted to the 4-bromo-2,5-dimethoxybenzonitrile (3) with acetic anhydride2.
Iodination of 1 using a similar procedure as for bromination was unsuccessful. Use of iodine monochloride had also been reported not to work3. Using stoichiometric amounts of iodine and silver nitrate in methanol4gave an 85% yield of 4-iodo-2,5-dimethoxybenzaldehyde (4). This was converted to 4-iodo-2,5-dimethoxybenzonitrile (5) in the same manner as the bromo compound.
Since bromination of 1 did not occur in good yield, 1 was converted to 2,5-dimethoxybenzonitrile (6), and bromination of 6 was investigated. Bromination in acetic acid did not occur in the 4-position to form 3, but rather in the 6-position to form (using correct numbering) 2-bromo-3,6-dimethoxybenzonitrile (7) in low yield. Exhaustive extraction of the reaction mixture did not yield more product. The 1H-NMR spectrum of 7 showed a pair of doublets with a 9 Hz coupling in the aromatic region, in contrast to the spectrum of 3, which showed a pair of singlets. Bromination using the procedure of Barfknecht and Nichols1 yielded a mixture of 3 and 7 by 1H-NMR.
Iodination of 6 yielded only 2-iodo-3,6-dimethoxybenzonitrile (8) in low yield, by either of two methods4,5. No 5 could be detected in the crude products by 1H-NMR.
In conclusion, 4-(bromo or iodo)-2,5-dimethoxybenzonitrile and 2-(bromo or iodo)-3,6-dimethoxybenzonitrile have been prepared by simple three-step procedures from 2,5-dimethoxybenzaldehyde. Although the overall yields are not high in some cases, pure products are readily obtained by recrystallization of the crude reaction products.
Experimental
4-Bromo-2,5-dimethoxybenzaldehyde (2)
To a solution of 5.0 g of 2,5-dimethoxybenzaldehyde (1) in 20 mL of acetic acid was added 1.8 mL of bromine
dropwise. The solution was allowed to stir for 4 hours. TLC (silica gel, ethyl acetate/hexane, 1:1) indicated
starting material was still present, so 1.0 mL of bromine was added, and the solution stirred overnight. The
reaction mixture was poured into water, and treated with 5% sodium bisulfite solution to remove the excess
bromine. The solid was filtered and washed with water. The solid was recrystallized from methanol to yield
3.45 g (46.9%) of 2, mp 122-4°C, lit.1 mp 132-3°C.
1H-NMR: 10.39 ppm (1H, s), 7.34 ppm (1H, s), 7.24 ppm (1H, s), 3.90 ppm (3H, s), 3.89 ppm (3H, s).
4-Bromo-2,5-dimethoxybenzonitrile (3)
Following the published procedure2, 2.0g of 4-Bromo-2,5-dimethoxybenzaldehyde (2) was converted to 1.69g (81.2%) of the oxime. Refluxing 1.07 g of the oxime in 5 mL of acetic anhydride for six hours, followed by dilution with ice water, suction filtration, and recrystallization from methanol, yielded 0.865g (76.6%) of 3, mp 175-7°C.
4-Iodo-2,5-dimethoxybenzaldehyde (4)
A mixture of 1 (5.10g, 30.7 mmol), silver nitrate (5.60g, 33 mmol), and iodine (8.10 g, 32 mmol) in 125 mL of
methanol was stirred under nitrogen for seven hours. The yellow precipitate was filtered and washed with methanol.
The filtrate was treated dropwise with just enough saturated sodium bisulfite solution to reduce the remaining iodine,
and the solvent was removed on a rotary evaporator. The solid was suspended with 50 mL of water, filtered, and
recrystallized from 95% ethanol to yield 7.62g (84.9%) of 4, mp 137-139°C, lit.3 mp 136-137°C.
1H-NMR: 10.39 ppm (1H, s), 7.47 ppm (1H, s), 7.22 ppm (1H, s), 3.90 ppm (3H, s), 3.88 ppm (3H, s).
4-Iodo-2,5-dimethoxybenzonitrile (5)
A solution of 4 (7.30g, 25 mmol) in 125 mL warm 95% ethanol was prepared on a steam bath. Hydroxylamine hydrochloride (2.08 g, 30 mmol) was dissolved in 15 mL of water, and added to the aldehyde solution. Sodium hydroxide (1.60 g, 40 mmol) was dissolved in 15 mL, of water, and added to the aldehyde solution. The solution was heated on the steam bath for 3 hours. The volume of solution was reduced to about 75 mL on the rotary evaporator, and the solution cooled to yield 3.97g of off-white crystals of the oxime. Further reduction of volume of the filtrate to about 40 mL yielded another 2.47g, for a total yield of 6.44g (84.0%) of the oxime. The oxime (5.0 g) was dissolved in 25 mL of acetic anhydride, and heated at reflux for 6 hours. The reaction mixture was cooled and diluted with water. The precipitated solid was filtered, washed with water, and recrystallized from methanol to yield 4.11g (87.3%) of 5, mp 153-5°C.
2,5-dimethoxybenzonitrile (6)
Following a published procedure2, 6 was prepared from 1 in 60% yield, mp 77-79°C (Lit.6 mp 82°C).
2-Bromo-3,6-dimethoxybenzonitrile (7)
To a mixture of 6 (3.50g, 21 mmol) in 15 mL warm acetic acid, bromine (1.2 mL, 24 mmol) was added dropwise with stirring. The reaction was stirred at room temperature overnight, poured into 80 mL of water, and the solid filtered by suction. The solid was recrystallized from methanol to yield 1.122g (22.1%) of 7, mp 150-152°C.
2-Iodo-3,6-dimethoxybenzonitrile (8)
Following a published procedure5, 6 (1.82g, 12.2 mmol), iodine (1.275g, 4.79 mmol), periodic acid (0.365g, 1.6 mmol), 0.5 mL of sulfuric acid, 15 mL of acetic acid, and 5 mL of water were stirred and heated at 60°C overnight. The reaction was cooled and diluted with water. The excess iodine was removed by treatment with sodium bisulfite solution, and the solid filtered, then recrystallized from methanol to yield 0.575g (17.7%) of 8, mp 167-169°C.
To a solution of 6 (1.0g, 6.0 mmol) in 50 mL of methylene chloride was added silver nitrate (2.04g, 12.0 mmol) and iodine (3.10g, 12.0 mmol). The solution was stirred under nitrogen overnight. The silver iodide was filtered, and the methylene chloride solution was washed with 2x50 mL of 5% sodium bisulfite solution and then with 50 mL of water. The methylene chloride was dried over magnesium sulfate, then removed under reduced pressure to yield a yellow solid, which was recrystallized from methanol to yield 0.58g (33.4%) of 8, mp 168-170°C.