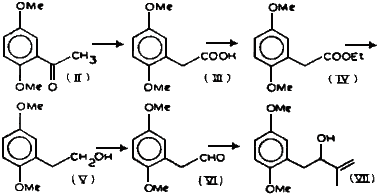
Summary
The ketone (II), prepared as reported1, was subjected to Willgerodt reaction2 to get the acid (III) which was changed into its ethyl ester (IV) in a high yield. LAH reduction of IV in ether gave the alcohol (V) which was oxidized to the aldehyde (VI) in 65% yield using pyridinium chlorochromate in dry methylene chloride3. The structure of VI was supported by its IR as well as NMR spectra. Aldehyde VI was subjected to the Grignard reaction with isopropenylmagnesium bromide to get alcohol VII.
Experimental
Melting and boiling points reported are uncorrected. IR spectra were recorded on a Perkin-Elmer model 337 spectrophotometer. The organic extracts were dried over anhyd. sodium sulphate.
2,5-Dimethoxyacetophenone (II)
II was prepared from dimethoxybenzene and acetyl chloride in the presence of anhydrous aluminium chloride according to the procedure described in the literature1.
2,5-Dimethoxyphenylacetic acid (III)
A mixture of II (45 g), sulphur (12 g) and morpholine (27 g) was heated under reflux for 6 hr and poured into ice-cold water. The crude reddish yellow mass obtained was thoroughly washed with water and to the crude thiomorpholide so obtained was added 500 ml of 10% ethanolic sodium hydroxide and the mixture refluxed for 10 hr. Most of the alcohol was removed, water (250 ml) added to the residue and the alkaline solution made strongly acidic. The contents were cooled and extracted with ether. The solvent was expelled from the ethereal extract to get the acid (III), mp 110?C, yield 24.5 g (50%); vmax 3300-2600 (broad, C-H and bonded OH), 1710 cm-1 (>C=O).
Ethyl 2,5-dimethoxyphenylacetate (IV)
A mixture of III (19.6 g), dry benzene (27.6 ml), dry ethanol (9.2 ml) and H2SO4 (2 drops) was refluxed under a Dean-Stark water separator until no water was collected in the limb (4 hr). After working up in the usual manner, the solvent was removed and the residue distilled under reduced pressure to give IV, bp 160-165?C/8-10 mmHg, yield 19 g (86%); vmax 1740 (ester carbonyl), 1600, 810 and 700 cm-1 (aromatic).
2-(2,5-Dimethoxyphenyl)ethanol (V)
To LAH (2.3 g) in dry ether (300 ml) was added dropwise the ester IV (16.8 g) in ether (60 ml) with stirring at room temperature. The stirring was continued further for 10 hr and the contents were cooled and decomposed-with saturated solution of sodium potassium tartrate. The ethereal layer was separated, the aqueous phase extracted with ether and the combined extracts dried. Removal of solvent and distillation of the residue gave V, bp 160-62?C/7-8 mmHg, yield 10.8 g (80%); vmax 3400 and 1050 cm-1.
2-(2,5-Dimethoxyphenyl)ethanal (VI)
Pyridinium chlorochromate (17.9 g) and sodium acetate (1.35 g) were suspended in 200 ml of dry dichloromethane and the alcohol (V, 10 g) in 50 ml of dichloromethane was added in one portion to the magnetically stirred solution kept at 10?C. After 2.5 hr, 200 ml of dry ether was added and the supernatant decanted from the black gum. The insoluble residue was washed thoroughly with anhyd. ether. The combined organic solution was passed through a short column of neutral alumina and the solvent removed. The crude product after distillation under reduced pressure afforded VI, b.p. 140-45?C/7-8mmHg, yield 6.5 g (65%); vmax 2720 (aldehydic C-H), 1715 cm-1 (-CHO); 60 MHz NMR spectrum (CCl4) exhibited signals at δ 3.5 (2H, d, J = 2 Hz, benzylic methylene), 3.7 and 3.8 (two s, 3H each for two OCH3), 6.6-7.1 (3H, Ar-H) and 9.65 (1H, t, J = 2 Hz, aldehydic proton)
2-Methyl-3-Hydroxy-4-(2,5-dimethoxyphenyl)but-1-ene (VII)
To the Grignard reagent, prepared from Mg (1.6 g) and isopropenyl bromide (8.2 g) in dry THF (250 ml) was added dropwise a solution of VI (6 g) in THF (20 ml). The reaction mixture was stirred for 2 hr and left overnight at room temperature. Thereafter it was decomposed with a saturated solution of ammonium chloride and worked up in the usual manner. The solvent was removed and the residue chromatographed over neutral alumina. Elution of the column with hexane-ether (1:1) furnished the carbinol (VII) as a thick liquid, yield 5 g (67.5%).