

#176 3-TSB
3-THIOSYMBESCALINE; 3-ETHOXY-5-ETHYLTHIO-4-METHOXYPHENETHYLAMINE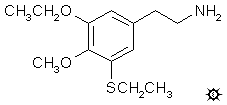
|
| [3D .mol structure] |
Several attempts were made to prepare the nitrostyrene from this aldehyde and nitromethane. The most successful, but still inadequate, procedure is described here. A solution of 1.0 g 3-ethoxy-5-ethylthio-4-methoxybenzaldehyde in 10 mL nitromethane was treated with about 150 mg of anhydrous ammonium acetate and heated on the steam bath. The course of the reaction was followed by TLC. The bulk of the aldehyde had disappeared in 45 min, and there were several UV-absorbing spots visible. Removal of the excess nitromethane under vacuum gave an orange oil which, when rubbed under cold MeOH, gave 200 mg of yellow solids. This was (by TLC) a mixture of nitrostyrene, starting aldehyde, and several slow-moving scrudge impurities. Recrystallization from MeOH gave a poor recovery of a yellow solid with a mp of 102.5-104 °C but this was still contaminated with the same impurities. Several repetitions of this synthetic procedure gave little if any of the desired 3-ethoxy-5-ethylthio-4-methoxy-beta-nitrostyrene.
A suspension of 5.4 g methyltriphenylphosphonium bromide in 30 mL anhydrous THF was placed under a He atmosphere, well stirred, and cooled with an external water bath. There was then added 10 mL of 1.6 N butyllithium in hexane which resulted in the generation of a bright pumpkin color. The initial heavy solids changed into a granular precipitate. There was then added 2.4 g of 3-ethoxy-5-ethylthio-4-methoxybenzaldehyde in a little THF. An initial gummy phase became granular with patient swirling and stirring. After 30 min, the reaction was quenched in 500 mL H2O, the top hexane layer separated, and the aqueous phase extracted with 2x75 mL of petroleum ether. The organic fractions were combined, washed with H2O, dried over anhydrous K2CO3, and the solvents removed under vacuum to give the crude 3-ethoxy-5-ethylthio-4-methoxystyrene as a yellow mobile liquid.
A solution of 2 mL of borane-methyl sulfide complex (10 M BH3 in methyl sulfide) in 20 mL THF was placed in a He atmosphere, cooled to 0 °C, treated with 4.2 mL of 2-methylbutene, and stirred for 1 h while returning to room temperature. To this there was added a solution of the impure 3-ethoxy-5-ethylthio-4-methoxystyrene in a little anhydrous THF. This was stirred for 1 h. The excess borane was destroyed with 1 mL MeOH, followed by the addition of 3.8 g elemental iodine, followed in turn by a solution of 0.8 g NaOH in hot MeOH added over the course of 5 min. The color gradually faded, and became a pale lime green. This was added to 300 mL dilute aqueous sodium thiosulfate which was extracted with 2x100 mL petroleum ether. The extracts were pooled, and the solvent evaporated under vacuum to provide crude 1-(3-ethoxy-5-ethylthio-4-methoxyphenyl)-2-iodoethane as a residue.
To this crude 1-(3-ethoxy-5-ethylthio-4-methoxyphenyl)-2-iodoethane there was added a solution of 3.7 g potassium phthalimide in 50 mL anhydrous DMF, and all was heated on the steam bath. The reaction seemed to be complete after 15 min (as seen by TLC) and the addition of a second batch of potassium phthalimide in DMF produced no further change. After adding to 500 mL of dilute NaOH, the aqueous phase was extracted with 2x75 mL Et2O. These extracts were combined, washed first with dilute NaOH and then with dilute H2SO4, dried over anhydrous K2CO3, and the solvent removed under vacuum which provided an amber oil as residue. This was triturated under cold MeOH giving white solids which were recrystallized from 20 mL MeOH. Thus there was obtained 0.9 g of 1-(3-ethoxy-5-ethylthio-4-methoxyphenyl)-2-phthalimidoethane as white crystals that melted at 79-80.5 °C. A small sample was recrystallized from EtOH to give large flat needles with a mp of 81-82 °C. Anal. (C21H23NO4S) C,H.
A suspension of 0.8 g of the crystallized 1-(3-ethoxy-5-ethylthio-4-methoxyphenyl)-2-phthalimidoethane in 25 mL of n-butanol was treated with 2 mL of 66% hydrazine, and the mixture was heated on the steam bath for 0.5 h. Initially all went into solution, and then there was the separation of solids that resembled cottage cheese. The reaction mixture was added to 150 mL dilute H2SO4. The solids were removed by filtration, and the filtrate was washed with 3x50 mL CH2Cl2. These washes were discarded. The H2O phase was then made basic with aqueous NaOH, extracted with 2x75 mL CH2Cl2, and the solvent from these pooled extracts removed under vacuum. The residue was distilled at 135-155 °C at 0.3 mm/Hg to give 0.45 g of a colorless oil. This was dissolved in 2.5 mL IPA, neutralized with 5 drops of concentrated HCl, and diluted with 10 mL anhydrous Et2O. The solution became cloudy, and then deposited lustrous white plates. These were removed by filtration, washed with additional Et2O, and air dried to give 0.4 g of 3-ethoxy-5-ethylthio-4-methoxyphenethylamine hydrochloride (3-TSB) with a mp of 153.5-154.5 °C. Anal. (C13H22ClNO2S) C,H.
DOSAGE: greater than 200 mg.
DURATION: unknown.
QUALITATIVE COMMENTS: (with 200 mg) No effects whatsoever, neither mental nor physical.
EXTENSIONS AND COMMENTARY: The elephant labored and brought forth a mouse. A lot of work for a material without activity.
I have used the term "scrudge" in this and other recipes, without defining it. With this aldehyde, as with most aldehydes in this nitrostyrene synthesis reaction where there is no ortho-substituent on the benzaldehyde, the reaction progress should be carefully followed by thin-layer chromatography. As the aldehyde disappears from the reaction mixture, the nitrostyrene appears, but there is usually the development of one or more slower moving components as seen by TLC. Such a wrong-product is called scrudge. The reaction should be continuously titrated, and stopped when there is a favorable balance between the aldehyde being mostly gone, the nitrostyrene being mostly made, and the slower-moving scrudge components being not yet too plentiful. Methylene chloride is an excellent solvent to try first, with silica gel plates and UV detection. The nitrostyrene is always the fastest moving component of the reaction mixture and often fluoresces a dull purple. The starting aldehyde is the second spot and usually fluoresces white or pale yellow. The scrudge spots then occur in a cascade from the aldehyde to the origin. A maddening property is that they are yellow or brown colored, and in the probe mass spectrograph they can crack to give rise to what appears to be the right nitrostyrene. Usually, they are high melting.
In this preparation, there was not one but several scrudges, and little if any nitrostyrene. The same was true for the other of the diethyl compounds such as 3-TASB, 5-TASB and 3-T-TRIS. Thus, it is preferable to circumvent this usual synthetic step by using the Wittig reaction instead, as described here.
| [ |
[Main Index] | [Forward |