

#132 MMDA
3-METHOXY-4,5-METHYLENEDIOXYAMPHETAMINE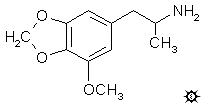
|
| [3D .mol structure] |
To a solution of 11.7 g 3-bromo-4,5-dihydroxybenzaldehyde in 36 mL DMSO there was added 29 g methylene iodide followed by 20.8 g anhydrous K2CO3. This was heated on the steam bath for 3 h, added to 1 L H2O, made strongly basic with NaOH, then extracted with 3x100 mL CH2Cl2. These extracts were pooled, washed with H2O, and the solvent removed under vacuum. The dark brown semi-solid residue was distilled with the major fraction (6.0 g) coming over at 120-130 °C at 0.3 mm/Hg. This, upon recrystallization from 35 g boiling MeOH, gave 1.3 g of 3-bromo-4,5-methylenedioxybenzaldehyde as an off white crystalline solid with a mp of 123-124 °C.
A mixture of 2.2 g 3-bromo-4,5-methylenedioxybenzaldehyde and 3.6 mL cyclohexylamine in a distillation flask was heated to 100 °C to effect solution, and then with an open flame until the signs of H2O evolution were evident. This was then placed under a hard vacuum to remove the generated water and excess cyclohexylamine, and the product distilled at 120-125 °C at 0.2 mm/Hg. There was obtained 2.4 g of the Schiff base of the aldehyde and the amine, melting at 86-96 °C. Recrystallization of an analytical sample from 5 volumes of MeOH gave 3-bromo-4,5-methylenedioxybenzylidine-N-cyclohexylamine as a white solid with a mp of 97.5-98.5 °C. Anal. (C14H16BrNO2) H; C: calcd, 54.20; found, 53.78.
A solution of 2.2 g 3-bromo-4,5-methylenedioxybenzylidine-N-cyclohexylamine (the above Schiff base) in 50 mL anhydrous Et2O was placed in a He atmosphere, stirred magnetically, and cooled with a dry ice/acetone bath. A white fine crystalline phase appeared. There was then added 5.2 mL 1.55 M butyllithium in hexane (the fine solids dissolved) followed by 4.0 mL of tributyl borate. After returning to room temperature, the reaction was quenched with 20 mL of saturated aqueous ammonium sulfate. The Et2O/hexane layer was separated, washed with additional ammonium sulfate solution, and then stripped of volatiles under vacuum. The residue was dissolved in 100 mL 50% MeOH, treated with 2 mL of 30% hydrogen peroxide and, after 15 min swirling, quenched with a solution of 10 g ammonium sulfate in 50 mL H2O. This aqueous phase (pH about 8) was extracted with 2x50 mL CH2Cl2, the extract pooled and stripped of solvent under vacuum, and the residue dissolved in warm, dilute HCl. After all the residue had dissolved (a few min heating was sufficient), the solution was cooled to room temperature and extracted with 2x50 mL CH2Cl2. These organics were pooled and extracted in turn with 2x50 mL 5% NaOH. Acidification of the pooled aqueous fractions with HCl, followed by extraction with 2x50 mL CH2Cl2 gave, after evaporation of the solvent, a residue that was distilled at 140-150 °C at 0.25 mm/Hg to give 3-hydroxy-4,5-methylenedioxybenzaldehyde. This was recrystallized from toluene (40 mL/g) to give 0.46 g of an off-white product with a mp of 134-134.5 °C. Anal. (C8H6O4) C,H.
A solution of 0.44 g 3-hydroxy-4,5-methylenedioxybenzaldehyde in 10 mL dry acetone was treated with 0.5 g methyl iodide and 0.5 g powdered anhydrous K2CO3, and was held at reflux for 6 h. All volatiles were stripped under vacuum, the residue dissolved in water, made strongly basic with NaOH, and extracted with 3x50 mL CH2Cl2. Removal of the solvent gave myristicinaldehyde (mp 133-134 °C) which, on recrystallization from hexane, gave a final yield of 0.42 g with a mp of 134-135 °C. Care must be taken with two sequential products that have identical mps. A mixed mp with the unmethylated phenol above is strong depressed, whereas that with an authentic sample is not.
A solution of 9.8 g myristicinaldehyde in 35 mL glacial acetic acid was treated with 5.3 mL nitroethane and 3.2 g anhydrous ammonium acetate, and heated on the steam bath for 1.5 h. It was removed, treated with H2O with good stirring to just short of turbidity, seeded with product nitrostyrene, and allowed to come slowly to room temperature. The bright yellow solids that formed were removed by filtration, washed with a small amount of aqueous acetic acid, and sucked as free of solvent as possible. This material, pressed on a porous plate, had a mp of 107-110 °C. Recrystallization from 60 mL boiling EtOH gave, after filtering and air drying, 5.1 g of 1-(3-methoxy-4,5-methylenedioxyphenyl)-2-nitropropene as light yellow solids with a mp of 109-110 °C.
A suspension of 7.5 g LAH in 500 mL anhydrous Et2O was magnetically stirred, and heated in an inert atmosphere to a gentle reflux. The condensing Et2O leached out a total of 9.8 g 1-(3-methoxy-4,5-methylenedioxyphenyl)-2-nitropropene from a Soxhlet thimble in a shunted reflux condenser. This, in effect, added the nitrostyrene to the reaction medium as a warm saturated Et2O solution. When the addition was completed, the refluxing was maintained for an additional 5 h, then the reaction mixture was cooled and the excess hydride destroyed by the addition of 400 mL 1.5 N H2SO4 (the first 20 mL a drop at a time and with very good stirring). The phases were separated, and sufficient saturated aqueous Na2CO3 was added to the aqueous phase to bring the pH up to about 6.0. This was heated to 80 °C and filtered through a coarse sintered glass funnel to remove some insoluble fines. The clear filtrate was brought up almost to a boil, and treated with a solution of 10.2 g of 90% picric acid in 110 mL boiling EtOH. Crystals of the picrate formed immediately at the edges, and as the reaction flask was cooled in an ice tub, the entire reaction set to a yellow mass of crystals. These were removed by filtration, washed sparingly with 80% EtOH, and air dried to give 14.0 g of the picrate salt of MMDA, with a mp of 182-184 °C. Recrystallization of a small sample from EtOH dropped this to 179-181 °C. This salt was treated with 30 mL 5% NaOH, and the red solution decanted from some insolubles. Additional H2O and NaOH effectively dissolved everything, and the resulting basic aqueous phase was extracted with 3x50 mL CH2Cl2. The pooled extracts were stripped of solvent under vacuum, and the residue dissolved in 200 mL anhydrous Et2O and saturated with anhydrous HCl gas. There was a heavy precipitation of white crystals, which were removed by filtration, Et2O washed, and air dried to give 6.37 g 3-methoxy-4,5-methylenedioxyamphetamine hydrochloride (MMDA) with a mp of 190-191 °C. Anal. (C11H16ClNO3) Cl.
(from Oil of Nutmeg) The careful distillation of Oil of Nutmeg (or the Oil of Mace) allowed the isolation of a number of compounds in varying degrees of purity. The fraction that boiled in the 110-115 °C range at about 1.0 mm/Hg was myristicin (3-methoxy-4,5-methylenedioxyallylbenzene). It constituted some 7% of the original oil of commerce and, in its original isolated form, was obtained with a purity of 87%. The major contaminant was elemicin (3,4,5-trimethoxyallylbenzene). A solution of 100 g myristicin in 100 g absolute EtOH was treated with 200 g solid KOH and heated on a steam bath overnight. Removal of the volatiles under vacuum, flooding the residue with H2O, and extraction with 3x100 mL CH2Cl2 gave, after removal of the solvent from the combined extracts, a residue of crude isomyristicin (a mixture of the cis- and trans-isomers). This product was distilled, and the fraction boiling at 125-130 °C at 1 mm/Hg gave 63 g of isomyristicin as a pale yellow oil that spontaneously crystallized. The mp was 41.5-42.5 °C. Part of the losses associated with the purification of these solids was due to formation of the cis-isomer of isomyristicin, which was an oil.
A solution of 50 g isomyristicin in 300 mL dry acetone containing 24 g pyridine was vigorously stirred and cooled to 0 °C with an ice bath. To this there was added 54 g tetranitromethane which had been pre-cooled to 0 °C. Stirring was continued for exactly 2 min, and then the reaction was quenched by the addition of a cold solution of 16.8 g KOH in 300 mL H2O. Stirring was continued until the temperature had again been lowered to near 0 °C. The product was removed by filtration. Extraction of the filtrate with CH2Cl2 and removal of the solvent provided additional nitrostryrene, for a combined yield of 50.7 g with a mp of 103 °C due to the presence of a small amount of free myristicinaldehyde. A recrystallization from MeOH produced 1-(3-methoxy-4,5-methylenedioxyphenyl)-2-nitropropene with a mp of 109-110 °C. This material was completely adequate for the above-described reduction to MMDA. The conversion of this nitropropene to myristicinaldehyde is an alternative to the lengthy synthesis given above), and can be used in the preparation of LOPHOPHINE.
A mixture of 50 g 1-(3-methoxy-4,5-methylenedioxyphenyl)-2-nitropropene and 26 g racemic a-methylbenzylamine was heated on the steam bath. The mixture gradually formed a clear solution with the steady evolution of nitroethane. When the reaction became quiet, there was added a mixture of 20 mL concentrated HCl in 100 mL H2O. The reaction mixture dissolved completely, and as the temperature continued to rise there was the abrupt solidification as the formed myristicinaldehyde crystallized out. This product was removed by filtration and, when combined with a second crop obtained by the hexane extraction of the filtrate, gave 36.9 g of myristicinaldehyde. The mp of 128-129 °C was raised to 133-134 °C by recrystallization from hexane.
DOSAGE: 100 - 250 mg.
DURATION: moderate.
QUALITATIVE COMMENTS: (with 100 mg) I felt completely relaxed at one hour. Almost as if I was floating. There were no obvious effects on taste, and the relaxation and composed feeling is much like a small dose, maybe 20 mikes, of LSD. There was some dilation, and in the evening I was a little restless and slightly tired. I slept well, and awoke refreshed and happy.
(with 100 mg) It seemed to take 45 minutes to work and then it came on very suddenly, as if my eyeballs were being pulled out and my whole head expanding. Soon a cold feeling set in with shivering--this was not unpleasant. My state in about two hours seemed to be one of empathy and passivity, compassion of an impersonal sort. The music sounded artificial and canned and tinny, in contrast to the voices, which sounded rich and full and finely articulated and melodious.
(with 150 mg) We are on the beach at the river mouth drying seaweed, on split redwood. There is a slight nausea, slight cramps, and then my visual field starts to light up. Still vertigo but only with my eyes open, and heaviness and time stretches out; numbness in the chest as when an opiate is taken. There are geometric patterns, but the excess light on my closed eyelids interferes with this. A dance of the glittering diamond studded sea waves, increasing motion and beauty. More landscapes appear inside. This is a good introductory drug to the drugs of this class, to become familiar with the drug state in as gentle a fashion as possible. This substance seems to have a much gentler action than others of this class; perhaps more like cannabis or psilocybin. There is very little paranoia. I note hallucinations of two types: those which are strictly retinal and more minute and small and influenced by light and focused on the light ahead on the retina or lids; and the other, those deep in the visual tract and occiput which are larger and more global and dream-like and, when solid, are quite dramatic and unforgettable as in meditation.
(with 210 mg) MMDA tastes awful. The bitter alkaloid taste is followed by a distinctively chemical laboratory flavor as if from old rubber tubing. Nothing seems to happen for about 45 minutes when rather suddenly an anvil seems to lower itself over your head; you feel disoriented, and tend to withdraw from social contact a little. The drug gives less feeling of being ill than mescaline. The effect definitely reaches a climax with a pleasant afterglow following. Apparently there are no profound motor coordination problems. MMDA yields that 'Sunday afternoon' feeling of desiring to lie down and enjoy life; a luxurious feeling of 'layback.' No enhancement of colors in visual scene (except for some greenish tinges in faces) but upon closing eyes hallucinations appear to be quite real in 3-D, like watching a movie. First these dreams appear in black and white, but later colors start appearing. Chartreuse and magenta first appear, then blue and finally red. First I had visions of large numbers on gaming tables, then people. MMDA appears to bring dreams to the conscious level; is a link between the subconscious and the conscious.
(with 225 mg) I had a strange awareness of my hands in about 20 minutes--not a feeling in them as just that I was attracted to them somehow. Then I began to get fearful, an acute experience of aloneness. I lay face down (a depressed position for me). Next I was talking to the kids at school (an image) or to other teachers. This was very vivid. The scenes at school were more vivid that the real scenes around me here. Those people were much more real. I am actually very sleepy right now during the experiment. Of any experience I have had, this was most like a series of dreams easily remembered. When it was over, I felt as if I had had a long period of sleeping--I had gone to bed and had a series of dream-like states very vivid and colorful and real.
EXTENSIONS AND COMMENTARY: The phrase that had been used by several of the subjects in the early trials with MMDA, again and again, was "brain movies." Apparently the richest of the effects were to be had with the eyes closed. This is the compound that I had first completed in 1962, and had named it MMDA, and had begun the exploring of it when I heard that Dr. Gordon A. Alles, a professor of pharmacology at U. C. L. A. who had his own private laboratory in Los Angeles, had also synthesized it in 1962, had also named it MMDA, and had also begun exploring it. We made a date to meet and share ideas, and then he died, at the age of 62, in 1963.
This is a material that might be a contributing factor to the pharmacology of nutmeg. The major essential oil from that spice is myristicin, and it is the easiest source of MMDA. It has been reported that the passage of this oil through the liver of a rabbit will generate MMDA in that animal. The only difference between the two molecules, structurally, are the elements of ammonia. Myristicin plus ammonia gives MMDA. Another natural source of myristicin is Oil of Parsley, which is also an excellent source of apiole, mentioned under DMMDA. A rumor that had currency in the 1960's, that parsley could get you high, probably had its origins in the reports of myristicin being present, coupled with myristicin being the principal source of MMDA. The relationship to myristicin (an essential oil) led to the classifying of MMDA as a Essential Amphetamine. These relationships are expanded upon, under TMA.
At the time that the FDA issued its proclamation of dangerous drugs (in the mid-1960's), MMDA was being talked about, and in fact it had just become available commercially in England through the Koch Light Industries. But to my knowledge it had never appeared on the street, so its having being swept into the listings of evil drugs was simply a coincidence of bad timing. The close resemblance of initials between MMDA, and the currently notorious MDMA, has led to no small amount of confusion in the popular press. They remain totally separate and completely different drugs.
| [ |
[Main Index] | [Forward |