
9 Physiological Effects of DXM
used at Erowid with the permission of author William WhiteThis section explains some of what is currently known about DXM and its physiological effects. As the recreational use of DXM is not well studied, most of that information is speculation, some of it entirely mine. To be more precise, a great deal of it is "scientific wild-assed guessing" (SWAGging) and should only be taken as interesting and unproven hypotheses.
A lot of this chapter is very technical. In the next chapter (Section 10), you will find an introduction to neuropharmacology, and the glossary (Section 16) contains definitions and explanations for the technical terms.
9.1 How Does DXM Inhibit the Cough Reflex?
This is a complex problem. The cough reflex generally involves a series of signals originating from the throat, lungs, and nasal passages, and ending up in the muscles. At any point in this pathway, signals can be blocked. Sigma receptors are evidently involved in this pathway (42,49,55,56). This may be a direct involvement - sigma activation may directly inhibit the cough reflex signals - or it may be an indirect one. The cough suppressant effect of opiates (such as codeine) is not related to the same effect of non-opiate morphinans like DXM (49); instead, it seems to be governed by traditional opiate receptors (mu, kappa, or delta).
There is some evidence that 5HT1A receptors (a serotonin receptor type) are involved somewhere in this pathway, and that cough suppressants may increase 5HT1A activity (57), possibly via NMDA antagonism (90). This could explain some of DXM's mood-altering activity. 5HT1A receptors are involved in anxiety states and in resilience to aversive events. Buspirone, a 5HT1A receptor partial agonist, is an anti-anxiety drug less potent (but considerably safer) than the benzodiazepines such as diazepam (ValiumTM).
9.2 How Does DXM Cause its Psychoactive Effects?
If you thought the cough reflex was complicated, just wait!
9.2.1 General Information
DXM binds to at least four sites in the brain (58), which can be arbitrarily labeled DM1, DM2, DM3, and DM4; there is probably also a fifth binding site (DM5). Some of these sites are sensitive to pentazocine, a known sigma ligand; some are sensitive to haloperidol, another sigma ligand. On the following table, information from several sources has been gathered and combined. The binding affinity of DXM, DTG, and 3-PPP are listed (58), along with (+)-pentazocine sensitivity (60), and haloperidol displacement ability (58) (binding values in nM unless otherwise specified). "Low" means micromolar binding affinity.
| DXM Site: | DM1 | DM2 | DM3 | DM4 |
|---|---|---|---|---|
| Probable Binding Site: | Sigma1 | PCP2 | Sigma2 | NMDA |
| DXM | 50-83 | 8-19 | low | low |
| (+)-3-PPP | 24-36 | low | 210-320 | low |
| DTG | 22-24 | ??? | 13-16 | ??? |
| Pentazocine Sensitive? | Yes | No | ??? | ??? |
| Haloperidol Displaced? | Yes | ??? | Yes | ??? |
What this table demonstrates is that DXM binds to four separate places, two with high affinity. The first receptor is accepted to be the sigma1 receptor based on the binding to pentazocine and haloperidol, and the potency of (+)-3-PPP. The second receptor is almost certainly the PCP2 receptor, given the insensitivity to pentazocine, and the very high affinity for DXM. The third site is probably sigma2 (based on the potency of DTG) but it is possible that "DM1" in this table represents both sigma1 and sigma2 and that the third site is something else. The fourth site is probably the NMDA receptor's open channel site, although it might be the ion channel binding site (59).
I don't have information on the binding of DXO (dextrorphan), unfortunately. It would probably show strongest binding at DM4 (NMDA open channel site), followed by DM1 (sigma1), and then by DM3 (sigma2) and/or DM2 (PCP2), or both. I'm looking for this information currently.
9.2.2 Contribution of the PCP2 Binding Site
The PCP2 binding site is probably the dopamine reuptake complex, so blocking it would prevent the uptake of dopamine in much the same way that the antidepressant bupropion (WellbutrinTM) or cocaine does (73). Of course, DXM is considerably weaker than cocaine (and stronger than bupropion, incidentally) at this site. This probably accounts for the euphoric effects of a low recreational dose, and almost certainly explains the stimulant effects of a low dose. Interestingly, the stimulant effect seems qualitatively different from amphetamines to most people (I have no comparison information on cocaine). One user compared DXM and bupropion favorably in stimulant effect. Incidentally, it seems that DXM may bind noncompetitively at the dopamine reuptake site whereas bupropion binds competitively.
The music euphoria and motion euphoria are probably partly due to PCP2 activity, and partly due to other activity. As NMDA blockade and sigma activity can both lead to dopaminergic activity (see below), reuptake inhibition would potentiate these effects.
Interestingly, DXM seems to be much more potent at this site than other sigma/NMDA ligands (such as PCP or ketamine) in comparison to activity at other sites. Also interestingly, at least one tricyclic antidepressant has been found to be active at related receptors (sigma, PCP) (71,74,75); it is possible that the PCP2 site may be a target of some antidepressants.
9.2.3 Contribution of the Sigma Binding Sites
As the sigma2 site is a fairly recent discovery, it is not known what sigma-related effects and behaviors are attributable to which receptor (sigma1 or sigma2). There is very little data on the subjective effects of sigma ligands, in part because only recently have selective ligands become available, and in part because most researchers aren't very willing to dose themselves to find out. DXM binds to the sigma1 receptor and is generally considered to be an agonist at this receptor. DXM is probably also an agonist (as opposed to an antagonist) at sigma2, though it is much weaker there.
The disruption of sensory processing may be due in part to sigma activation (and partly due to NMDA blockade) (63-65). Sigma receptors may be specifically involved in the auditory effects of DXM (65), and these effects may relate to a disruption of sensory input persistence.
The psychotomimetic (literally "psychosis-like") effects of DXM may be a result of sigma activity since sigma receptors seem to have some involvement in schizophrenia (46-49). People who have used both DXM and ketamine have remarked that DXM is much more likely to induce delusional and hyper-abstract thought patterns. Sigma receptors may temporarily modulate cholinergic receptors (97), so sigma activity may produce temporary effects somewhat like the delusional anticholinergics.
The effects on motor skills may be a result specifically of sigma2 receptors (69). Expect to see more data on this subject as sigma2 receptors are investigated more fully. There may also be a contribution from NMDA receptors, of course.
9.2.4 Contribution of the NMDA Receptor
Dextrorphan (DXO) is much more potent than DXM at the NMDA receptor, which accounts for the slow onset of most of DXM's effects even when it is injected. The NMDA receptor is the central site of dissociative action, and is probably the main contributor to most of DXM's effects. Dissociatives, including DXM, act on the NMDA receptor by binding to its channel once it opens up, essentially plugging it up.
Most of the "stoning" or intoxicating effects of DXM are due to NMDA receptor blockade. Alcohol's intoxicating effect seems to be mediated in part by NMDA receptor blockade (alcohol's depressant effect is due to GABA activity; DXM has no activity at GABA receptors) (28,61,62). The dissociative anesthesia of high DXM doses is also likely due to NMDA receptor blockade (63).
As stated before, sensory processing disruption, especially at higher doses, is probably due in part to NMDA receptors and partly to sigma (63-65). NMDA blockade is probably responsible for most if not all of the flanging effects of DXM, especially visual flanging. This is likely an indirect effect, i.e., the blockade of NMDA receptors induces changes in the way the brain processes information, leading to flanging.
The effects on memory are almost certainly due to NMDA blockade. NMDA receptors are intimately involved in long-term potentiation (64,66-68), the primary mechanism behind intermediate-term memory (ITM) and long-term memory (LTM). By blocking NMDA receptors, long-term potentiation, and thus intermediate- and long-term memory encoding, are disrupted.
NMDA blockade indirectly (244) increases dopamine activity in the striatum, nucleus accumbens, olfactory tubercule, and prefrontal cortex (204,226,326). Increased activity at dopamine D1 receptors is responsible for the increased locomotor activity seen in rats on dissociatives (178), and may be responsible for many of the effects DXM has on motion. Chronic use of DXM may result in upregulation of dopamine D2 receptors (205).
DXM's ability to suppress respiration at toxic levels is most likely due to NMDA receptor blockade or (in my opinion) ion channel blockade. Some of the effects from very high dosage levels may be due to overall disruption of neural networks. There is some preliminary evidence that both the "spontaneous memory" effect and the sensations similar to near-death experiences may occur as limbic areas (the hippocampus and hippocampal formation and surrounding areas) are disrupted by NMDA blockade. Most drugs target specific, small clusters of neurons (or their receptors, which can be scattered about). Both excitatory amino acid secreting neurons and NMDA receptors tend to be more evenly distributed, although they too are concentrated in certain areas of the brain to a lesser extent.
9.2.5 Temporal Lobe Involvement
Probably the greatest degree of DXM's effects come from consequences of the aforementioned receptor binding on the temporal lobe limbic areas. The blockade of NMDA receptors by DXM has specific consequences in these areas. The normal functioning of the hippocampus and amygdala are disrupted since NMDA blockade prevents long-term potentiation. The posterior cingulate and retrosplenial cortex paradoxically become more active. There may be spontaneous "noise" effects in the entire limbic system amplified by feedback. One paper suggested that dissociatives may induce "microseizures" or limited areas of high activity in the limbic system (176).
There is also the suggestion of a "top-down" inhibition of the senses, i.e., inhibitory signals from the limbic areas and surrounding cortical areas are sent "down" to sensory networks, lowering the strength of sensory data. This "top-down" inhibition of the senses may be the mechanism behind dissociative anaesthesia; sensory information is still processed by the brain, but never makes it to the conscious mind and is never encoded in intermediate-term declarative memory (in the hippocampus).
Finally, dissociatives seem to alter the flow of signals through the limbic areas, possibly increasing the degree of internal feedback within these areas (or between these areas and the neocortex) and diminshing the amount of sensory data that comes in. Gating of signals coupled to the theta rhythm through the posterior cingulate (311) may be altered.
Putting this all together into some sort of cohesive theory may be too soon but I'm going to do it anyway. My belief (which will probably change as new research becomes available) is that the diminished sensory data (from top-down inhibition), and the decreased encoding of intermediate-term memory, combined with the enhancement of activity in the posterior cingulate and retrosplenial cortex, all lead to an increasingly closed feedback loop. Within this loop, random noise, individual differences in temporal lobe "wiring", the contents of intermediate memory, and the influence of electromagnetic fields (332) all combine to give rise to profoundly abnormal neural patterns.
An interesting aside to this is that the sense of smell is sometimes reported to be enhanced on DXM. This hasn't been formally studied, and may be all in one's mind, but since olfactory data is treated somewhat differently by the brain than the other senses, it may be spared from the descending inhibitory signals that attenuate other sensory data. If so, then the feedback loop could actually serve to increase the strength of olfactory data, by repeatedly adding the same, small signals together.
9.2.6 Contributions of Indirect Activity
Many of DXM's effects are undoubtedly due to indirect activity at other neurotransmitter systems. For example, it may indirectly increase 5HT activity, especially at the 5HT1A receptor. This could explain some of its mood-altering properties. Another example is dopaminergic activity; DXM has a fairly strong ability to increase dopamine activity (both from activating sigma receptors, and from preventing dopamine reuptake at PCP2 sites) (72,76). NMDA receptor blockade also has been shown to increase dopaminergic activity, as well as activity of other neurotransmitter systems (100).
9.2.7 Flanging
One of DXM's most prominent effects if the flanging of sensory input. This happens to some extent with many drugs, and I have a hypothesis on this. Note in particular the relation of flanging to "stoning" and "buzzing" - in some ways, flanging is a more profound degree of stoning. Some people have noticed a flanging or strobing effect after smoking a great deal of cannabis, and nitrous oxide users are also familiar with flanging of sounds. Even alcohol can produce it.
What it seems many of these drugs have in common is the ability to inhibit hippocampal long-term potentiation. Some have suggested that there are more than one set of signals that pass through the same limbic areas, with those in phase with the theta frequency and those out of phase being gated differently. Perhaps one set of signals is more strongly involved with memory, and the other set more strongly involved with sensory data. Or, perhaps all drugs which inhibit hippocampal LTP all activate the posterior cingulate cortex. In either case, signals at one phase of the theta rhythm could be disrupted, leading to a pulsing of perception in step with theta rhythm.
A different theory is that networks in the brain will re-process the same data repeatedly until a stable configuration is formed, and that DXM (and other drugs) slow down this process. With some networks slowed down and others operating at normal speeds, the characteristic frequencies of the two sets of signals would differ, leading to a "beat frequency" in much the same way that two very similar sounds can lead to a beat frequency (for a quick demonstration of this, listen to someone tuning a guitar by ear).
9.2.8 Hyper-Abstraction
Another interesting effect of DXM is its ability to induce peculiar cognitive disturbances, which I lump together under the term of "hyper-abstraction". Two examples:
- A meme is a "particle" or "virus" of thought - an idea which is in some ways self-contained, and which spreads like a virus. For example, the idea of civil liberties is a meme, which at some point sprang into existence, spread rapidly, and has now become an integral part of our consciousness. One user during a DXM trip suddenly became aware of (or thought up) "The self-invoking, self-creating meme", which was the concept of a meme whose identification creates and invokes it. It seemed that this meme was timeless in the sense that it must have always existed, or it could not have come to mind, since it is not easily deducible from anything other than itself (of course, the brain doesn't work on the rules of logic, but ... you get the point)
- Another user wrote of thinking about convergent infinite sums (e.g., 1/2 + 1/4 + 1/8 + etc., which sums up to 1). Although one can add these terms up forever, it's easier to abstract the process and get the answer that way. This user imagined an infinite series of abstractions, and then imagined abstracting that infinite series to get a new level or plane of abstraction.
Many DXM thought patterns involve what some have called "Strange Loops" in logic (137). Like the self-contradicting statement "this statement is false", some of them cannot be embodied in logical form; others can be, but cannot be derived without presenting them as hypothesis. Thinking at this degree of abstraction is very difficult (unless you are fortunate enough to be Kurt Gödel).
Several people who have written first-person accounts of psychosis and schizophrenia have mentioned increasingly abstract thought patterns (Zen and the Art of Motorcycle Maintenance springs to mind). This may of course be complete bunk, and it may be that the increasingly "abstract" thoughts are just increasingly loony (and thus difficult to relate to concrete ideas). On the other hand, it may be that something about schizophrenia and psychotic states is related to a blurring between levels of abstraction. Once blurred sufficiently, a thought which cannot be represented at a concrete degree of abstraction could be representable in the mind.
Thus, DXM may induce a sort of temporary blurring of these levels of abstraction. Whether this is due to NMDA or sigma activity, I don't know, although I suspect the latter, since other NMDA antagonists don't tend to induce such changes in thought patterns.
9.2.9 Delusions and Memory Problems
As stated above, sigma activity may modulate cholinergic receptors in the brain (97), leading to a temporary decrease in cholinergic function similar to (but considerably safer than) that caused by anticholinergics like atropine, scopolamine, cyclizine (Marezine), etc. It is known that cholinergic activity is important in memory, and many nootropics ("Smart Drugs") enhance cholinergic function. Sigma activity may very well cause temporarily lowered effectiveness in some cholinergic receptors, thus distorting memory and thought processes. Some people have in fact said that DXM makes them feel temporarily stupid ("Dumb Drugs", anyone?) although this by no means happens to everyone.
Many of the biogenic amine systems seem to have a modulatory role, and some researchers think these modulating systems operate much like "control knobs". For example, one theory on LSD is that it upsets the "gain control" on sensory recognition networks (possibly by descending inhibition, although through different pathways than those postulated for dissociatives). As a consequence, the random noise input (necessary for any pattern matching network) becomes much stronger than the sensory input. Sensory recognition becomes increasingly less and less precise - ergo, hallucinations.
LSD's effects are almost certainly more complex than this, but there may be some truth to the "control knob" idea of the biogenic amine systems. If so, then the cholinergic systems may be the "control knobs" for cognitive networks in much the same way that 5HT2A/5HT2C systems are for sensory recognition networks. Delusions may simply be the cognitive equivalent of hallucinations. Or to put it another way, the difference between thinking you look like a flower and thinking you are a flower may be a question of which network is disrupted.
Memory problems derive to some extent from NMDA blockade, although some users of ketamine have remarked that DXM can have a stronger effect on memory than ketamine. It is possible that, in addition to inducing delusional thoughts, a decrease in cholinergic function could be responsible for some of the memory problems. This is certainly consistent with the effects of the delusional anticholinergics.
Incidentally, the anticholinergics also affect acetylcholine receptors that govern the functioning of the heart and respiration (these receptors do not seem to be modulated by sigma activity). Recreational use of anticholinergics can be extremely dangerous, leading to collapse of respiration or heart failure.
9.3 Why Does DXM Exhibit Plateaus?
9.3.1 Plateaus 1-3: Multiple Receptors
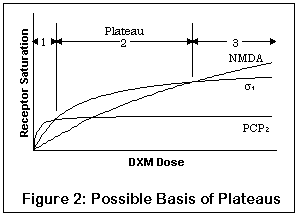 This graph illustrates a potential effect of multiple receptors.
Note that it is a qualitative drawing, not a quantitative one;
the actual degree of saturation on different receptors for a given
strength of DXM is still mostly unknown. The lines represent
saturation levels at the PCP2, sigma1, and NMDA
open channel sites,
with full saturation at a given receptor indicated by the "flattening
out" of the curve. The X axis is the concentration of DXM; the Y
axis is the percent saturation of receptors.
This graph illustrates a potential effect of multiple receptors.
Note that it is a qualitative drawing, not a quantitative one;
the actual degree of saturation on different receptors for a given
strength of DXM is still mostly unknown. The lines represent
saturation levels at the PCP2, sigma1, and NMDA
open channel sites,
with full saturation at a given receptor indicated by the "flattening
out" of the curve. The X axis is the concentration of DXM; the Y
axis is the percent saturation of receptors.
Incidentally, if you are familiar with this sort of thing, you may be more comfortable with a Lineweaver-Burk plot, but for those unfamiliar with them they can be confusing.
Due to its increasing affinity for PCP2, sigma1, and NMDA receptors respectively (sigma2 is not represented), a low dose will tend to have proportionally more effect on PCP2 receptors, whereas as the dosage increases, these receptors will saturate. Taking more DXM won't change PCP2 levels much, but will still have a fair effect on other receptors.
Furthermore, the more subtle effects on the PCP2 receptors may be all but obliterated by the effects on sigma1 and NMDA receptors (the differing vertical maxima of the three curves represent this effect). This is entirely reasonable, since sigma1 and NMDA activity seem to both produce fairly profound behavioral effects, the latter more so than the former.
Thus, the first plateau may correspond to predominantly PCP2 activity with some sigma activity and a little NMDA blocking effect; the second plateau to sigma and some NMDA effects; and the third to profound NMDA blockade.
This is, of course, a simplification, and it certainly doesn't take into account individual variations in receptors. Some people probably have genetic variants of receptors for which DXM has a stronger (or weaker) affinity. A person with PCP2 receptors strongly bound by DXM may enjoy its stimulant effects a great deal more than someone whose PCP2 receptors are only weakly affected.
Additionally, both ion channels and sigma2 receptors are omitted from this graph. They undoubtedly contribute, and some people have asserted that there are plateaus in between the first three. Some of these may be so subtle as to be unnoticeable by most users.
9.3.2 The Fourth Plateau: Sensory Shutdown
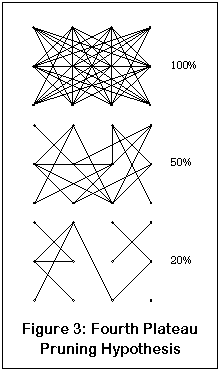
What about the fourth plateau? Well, once again, here's another little drawing that will hopefully clear up everything (yeah, right). This drawing represents a neural network; the dots are the neurons and the lines are their connections. Like most of the brain, this network is highly interconnected. The percentage numbers show the number of functional links remaining.
As enough NMDA receptors are blocked, one neuron may lose enough input from another that the connection is effectively severed. Initially this isn't such a problem, since both neurons and connections are dense enough so that others can take over the job (although the end result will probably be a slower and less accurate network). At some point, enough connectivity is lost that the network no longer functions.
Compare this to the dissociation of the fourth plateau. At some level, some part of the brain (possibly the hippocampus) loses enough functionality that it can no longer operate as a cohesive unit. Sensory processing halts, and raw sensory input cannot be converted to an appropriately parsed output. The consciousness is then left without any real sensory input; instead, the chaotic, unstable patterns are provided. Ergo, dissociative anesthesia.
On the other hand, it may be that the threshold of the Fourth Plateau is reached when sensory information becomes inhibited enough for the neural signals in the limbic areas to feed back without substantially being "grounded" in sensory input. This feedback loop continues to perturb the neural signal patterns until they are totally disconnected from any state one is likely to encounter from either sensory input or memory.
9.4 Why is This So Complicated?
DXM itself is a very complex drug; most drugs only bind to one or two receptors (or at least one class of receptors). Its recreational abuse potential, although known for years, has not been well studied, and it can affect different people very differently. The receptors and binding sites it affects - sigma, NMDA, and PCP2 - are all new discoveries. All this adds up to a complicated and poorly understood drug.
Furthermore, the brain itself is a complicated system, and we're still mostly ignorant of its function. The basics of neurotransmission seem to be understood, but many questions remain. Nobody knows why there are so many different neurotransmitters, nor why there are so many receptor subtypes. The second messenger systems of most receptors are not well understood either. A lot of what happens inside neurons occurs via changes in genetic expression, and that's yet another topic about which little is known.
To repeat a commonly quoted (and true) sentiment, if our brains were simple enough for us to easily understand, we would be so simple that we couldn't understand them. I do believe that eventually we will have a good idea of how the brain works, but it may not be in my lifetime. Until then we're all just making guesses in the dark.
9.5 Pharmacokinetics: How DXM is Metabolized
DXM, as the hydrobromide salt, is absorbed quickly from the GI tract; within 30 minutes, all of it may have entered the bloodstream (2,3). The polistirex compound is intended for continuous absorption, and may take 6 to 8 hours to fully enter the bloodstream.
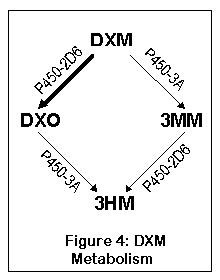
DXM is metabolized via two pathways, both of which lead to the same thing, 3-hydroxymorphinan (3HM). The first pathway goes from DXM to DXO (dextrorphan) and then to 3HM; the second goes from DXO to 3-methoxymorphinan (3MM) and then to 3HM. By far most of the DXM (up to 90%) gets metabolized via DXO in normal individuals.
DXM is converted to DXO by a liver enzyme called cytochrome P450-2D6 (debrisoquine 4-hydroxylase). About 7% of Caucasians and 0.5% of Asians have a highly inefficient (70 times slower) version of this enzyme, and cannot metabolize DXM to DXO effectively (10). About 0.5 to 2% of the population have multiple copies of the P450 gene, leading to extremely fast metabolism of DXM into DXO (155). After being converted to DXO, the enzymes P450-3A4 and P450-3A5 convert DXO to 3-hydroxymorphinan (77).
The other pathway goes to 3-methoxymorphinan first (via P450-3A4 and P450-3A5), and then to 3-hydroxymorphinan. Most people do not metabolize much DXM this way, although people who lack the normal P450-2D6 will convert a substantial amount to 3MM. As 3MM is probably not psychoactive, this means that the 5-10% who lack the normal 2D6 enzyme will experience less effect from DXM (or more specifically, won't experience the effects of DXO).
P450-2D6 functions by removing the methoxy group and replacing it with a hydroxyl (OH) (or more accurately, pruning the methyl off the oxygen); this step is known as O-demethylation. P450-3A4 and 3A5 replace the methyl group with a hydrogen (H); this is the N-demethylation step. Refer to the diagram of the DXM molecule in Section 4.2 for the location of the methyl and methoxy groups.
9.5.1 Factors Affecting Metabolism of DXM
As stated above, some people lack the normal P450-2D6 enzyme. In the rest of the population, this enzyme can be inhibited by several factors. Many drugs inhibit P450-2D6, notably including fluoxetine (ProzacTM). A partial list of P450-2D6 inhibiting drugs is given in Section 15.1.
DXM itself naturally will compete with other drugs for P450-2D6, and importantly, so will 3-methoxymorphinan (3MM) (17). In fact, 3MM may have more affinity for the P450-2D6 enzyme than DXM itself does. This may account for the fact that a second "booster" dose of DXM generally produces different effects than the first dose; the competition for P450-2D6 will reduce the amount of DXM converted to DXO in the second dose.
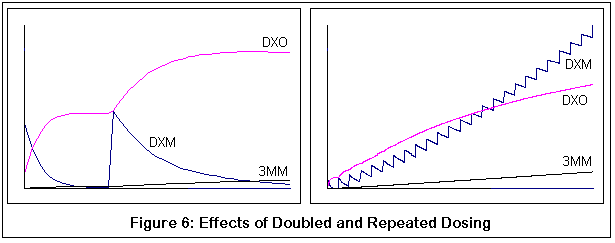
The following graphs come from computer simulations of DXM metabolism:
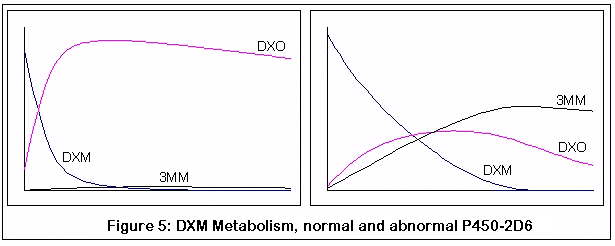
The first pair represent the metabolism of DXM in a normal individual (on the left) and an individual without the normal P450-2D6 enzyme (on the right). Note the rapid and almost complete conversion of DXM to DXO in the normal individual, as opposed to the less efficient and slower conversion in the P450-2D6 lacking individual.
The next pair demonstrate the probable results of taking additional doses (dose boosting). Both graphs correspond to individuals with the normal P450-2D6 variant. Note how the second dose of DXM is not converted to DXO as quickly (thus the shallower slope). The right hand graph shows numerous doses, and the "flattening out" of the metabolism curve for DXM is increasingly evident with each dose.
Incidentally, that these are qualitative simulations, not quantitative ones. I have tried to adhere to known KM and VMAX values for the applicable reactions, but the simulation was just a discrete process (to be honest, my differential equation skills are rusty enough that if you stepped on them you'd need a tetanus shot). I did compare my results with what little data I could find, and the comparison seemed reasonable, but then again I could be completely off base. The purpose of these graphs is to demonstrate the relative effects of changes in enzyme activity (via genetic variance and competitive inhibition by 3MM), and hopefully this is good enough for that purpose.
I have no information on what happens to 3-hydroxymorphinan itself. It may be excreted directly by the kidneys, or it may undergo further metabolism.

| 
| 
|