Synthesis of Nitrobenzene and Aniline[ Back to the Chemistry Archive ] Synthesis of nitrobenzene [1]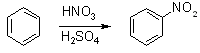 Place 50 g (35 mL, c. 0.5 mol)of concentrated nitric acid in a 500-mL round-bottomed flask, and add, in portions with shaking, 74 g (40 mL) of concentrated sulfuric acid. Keep the mixture cool during the addition by immersing the flak in cold water. Place a thermometer (110 °C range) in the acid mixture. Introduce 26 g (30 mL, 0.33 mol of benzene (CAUTION) in portions of 2 - 3 mL; shake the flask well to ensure thorough mixing, after each addition of benzene. Do not allow the temperature to rise over 55 °C; immerse the flask, if necessary, in cold water or in ice water. When all the benzene has been added, fit a reflux condenser to the flask and heat it in a water bath maintained at 60 °C (but not appreciably higher) for 40 - 45 minutes; remove the flask from time to time and shake it vigorously to ensure good mixing of the immiscible layers. Pour the contents of the flask into about 500 mL of cold water in a beaker, stir the mixture well in order to wash out as much acid as possible from the nitrobenzene and allow to stand. When the nitrobenzene has settled to the bottom, pour of the acid liquor as completely as possible, and transfer the residual liquid to a separatory funnel. Run off the lower layer of nitrobenzene and reject the upper aqueous layer; return the nitrobenzene to the separatory funnel and shake it vigorously with about 50 mL of water. Separate the nitrobenzene as completely as possible and rin it into a small conical flask containing about 5 g of anhydrous calcium chloride. If the nitrobenzene does not become clear because of the presence of emulsified water, warm the mixture, with shaking, for a short period on a water bath; the cloudiness will soon disappear. Filter the cold product through a small fluted filter paper into a small (50- or 100-mL)distilling flask and attach a still-head and air condenser. Heat the flask on a ceramic-centred wire gauze or preferably in an air bath, and collect the fraction which boils at 206 - 211 °C. (1). Do not distill quite to dryness nor allow the temperature to rise above 214 °C, for there may be a residue of m-dinitrobenzene or higher nitro compounds and an explosion may result. The yield of nitrobenzene is 35 g (85 %). Pure nitrobenzene is a clear, pale yellow liquid, B.P. = 210 °C.
Nitrobenzene Synthesis (alternative)Prepare a mixture of 82 mL of 95-100% sulfuric acid and 71 mL of 70% nitric acid in a 500-mL flask. Stir well and allow the mixture to cool to room temperature in a cold water bath. Gradually add 57 mL of benzene to the acid with frequent shaking. If the temperature rises above 50-60 °C during the benzene addition, stop adding benzene and cool the flask in a cold water bath until the temperature has lowered. After all of the benzene has been added, reflux the flask in a water bath at 60 °C for 1 hour. The temperature of the water bath should be 60 °C, not the contents of the flask. Shake the flask frequently during reflux. After heating, allow the flask to cool, two layers should form. Transfer the contents to a separatory funnel and drain off the bottom layer of sulfuric and nitric acids; the top layer contains the nitrobenzene. The bottom layer can be disposed of. The nitrobenzene is then vigorously shaken in the separatory funnel several times with water. After each shaking, allow the layers to separate, the nitrobenzene will now be the bottom layer, dispose of the top water layer. After washing, place the nitrobenzene in a dry Erlenmeyer flask with some calcium chloride. Heat this flask on a steam bath, it will first be milky, then it will go clear, stop when it is clear. The nitrobenzene is now purified by simple distillation. Yield is about 60-70 g. Synthesis of aniline [2]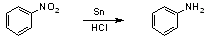 Into a 500-mL round bottomed flask equipped with a reflux condenser place 25 g (21 mL, 0.25 mol) of nitrobenzene and 45 g (0.38 mol) of granulated tin. Measure out 100 mL of concentrated hydrochloric acid. Pour about 15 mL of this acid down the condenser and shake the contents of the flask steadily. The mixture becomes warm and before long the reaction should be quite vigorous; if it boils very vigorously, moderate the reduction somewhat by temporarily immersing the flask in cold water. When the initial reaction slackens of its own accord, pour another 15 mL of hydrochloric acid down the condenser, shake the flask steadily to ensure thorough mixing and cool again if the reduction becomes too violent. Do not cool more than is necessary to keep the reaction under control; keep the mixture well shaken. Proceed in this way until all 100 mL of acid has been added. Finally heat the mixture on a boiling water bath for 30 - 60 minutes, i.e. until the odour of nitrobenzene is no longer perceptible and a few drops of the reaction mixture when diluted with water yield a perfectly clear solution. During the course of the reduction, particularly during the cooling, aniline chlorostannate may separate as a white or yellow crystalline complex. Cool the reaction mixture to room temperature and add gradually a solution of 75 g of sodium hydroxide in 125 mL of water; if the reaction mixture boils during the addition of alkali, cool again. The hydroxide of tin which is first precipitated should all dissolve and the solution should be strongly alkaline: the aniline separates as an oil. Equip the flask for steam distillation, and pass steam into the warm mixture until, after the distillate has ceased to pass over as a turbid liquid, a further 120 mL of clear liquid are collected. Since aniline is appreciably soluble (c. 3 %) in water, it must be 'salted out' by saturating the distillate with salt. Use about 20 g of commercial salt for each 100 mL of liquid. Transfer the distillate, saturated with salt, to a separatory funnel, add about 40 mL of ether and shake to ensure intimate mixing of the solution and the ether; relive the pressure within the funnel by momentarily lifting the stopper. (All flames in the vicinity must be extinguished during the extraction ... :-) you must be joking ...). Allow the two layers to separate; run off the lower aqueous layer into a beaker, and pour the ethereal layer through the mounth of the funnel into a 200-mL flask. Return the aqueous solution to the funnel and extract with a further 40 mL of ether. Proceed as before, and pour the ethereal extract into the flask. Dry the combined ethereal solutions with a few grams of anhydrous potassium carbonate (1): shake the well-stoppered flask for several minutes. Filter the ethereal solution through a fluted filter paper and remove the ether by flash distillation, using a 50-ml round-bottomed flask to which has been added a few boiling chips. Since ether is extremely volatile and also highly flammable, the flask must be heated by means of an electrically heated water bath. When all the ethereal solution has been introduced into the flask, and no more ether distilson the boiling water bath, run out the water from the condenser, and distil the aniline either by direct heating over a wire gauze or, preferably, using an air bath. A small quantity of ether may pass over during the early part of the distillation; it is therefore advisable to interpose a uralite board between the receiver and the flame. Collect the fraction B.P. 180 - 184 °C, in a weighed conical flask. The yield of aniline is 18 g (97 %).
References[1] Vogel, Practical Organic Chemistry, 5 th ed., p
854 |