Abstract
A re-examination of the methylenation reaction of 1-hydroxy-2-mercapto-, 1,2-dihydroxy- and 1,2-dimercapto substituted benzenes by bromochloromethane with cesium carbonate shows that these substrates give mixtures of five- and ten-membered benzocondensed heterocyclic compounds and in some cases even dibenzodioxines.
In previous papers we reported a systematic study on bimetallation of aromatics promoted by the thioethereal group. Recently, this research has been extended to 1,3-benzodioxoles and 1,3-benzoxathioles1,2 because these systems are contained in natural compounds, in bioactive molecules and in materials showing non-linear optical properties3-8.
It is known that these compounds can be prepared condensing 1,2-benzodioles or 2-hydroxybenzenethioles with dichloromethane or dibromomethane in the presence of alkaline hydroxides or fluorides or potassium carbonate or sodium hydride in various solvents (DMSO, DMF, HMPA) with or without catalytic amounts of bronze, nickel oxide or cupric oxide9-15. Good results can be obtained using phase transfer catalysts16,17. More recently, the methylenation of a variety of catechols has been reported, employing cesium carbonate and bromochloromethane in N,N-dimethylformamide or acetonitrile18.
In this work we intend to verify if this method can be applied to the methylenation of 2-hydroxybenzenethiol (1a).
All reactions were performed in anhydrous N,N-dimethylformamide reacting 1a, cesium carbonate and bromochloromethane. Reaction yields were determined by HPLC analysis of the reaction mixture obtained after extraction with solvents. The products were isolated as pure by flash chromatography (Scheme 1).
Surprisingly, the expected 1,3-benzoxathiole (2a), liquid at room temperature17, was obtained in a 32% yield, while the main product (65% yield) was a solid with mp 148–150°C: its elemental analysis was equal to 2a, but its mass spectrum showed a molecular ion M+ of 276, corresponding to the dimer 3a. Furthermore, the 1H-NMR spectrum of 2a showed the methylene singlet at δ 5.5017, while the solid product showed two singlets of equal intensity at δ 4.12 and 5.89 attributable to the two methylenes in 3a.
For a better understanding of this methylenation reaction, we decided to re-examine the reactions with catecholic derivatives. Using 1,2-benzodiol and its derivatives the reaction gave a mixture of benzodioxole and tetraoxecine derivatives. In detail, methyl derivatives 1c and 1d gave a significant amount of tetraoxecines ranging between 26 and 30%: in fact 1c gave a 1.1:1 mixture of two isomeric tetraoxecines 3c and 3’c; while 1d gave an almost equimolar mixture of 3d and 3’d. Employing the methoxy derivative 1g, we obtained a mixture of the benzodioxole 2g (68%) and the isomeric tetraoxecine 3g and 3’g (17%) beside two isomeric dibenzodioxines 4 (or 4’) and 5 (or 5’), in almost equal amounts (Fig. 1).
Then we decided to apply this reaction protocol to 1,2-dimercaptobenzene (1h): 1,3-benzodithiole (2h) (98%) was obtained almost exclusively beside traces amount of unknown products.
The same reactions performed in acetonitrile and at different concentrations gave analogous results. At the end we can deduce that when the benzene ring bears two equally nucleophilic groups (benzodioles and dimercaptobenzenes) the main product is the one with a five-membered ring; when the groups have a different nucleophilic power the main product is the ten-membered ring.
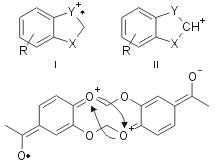
The structure determination of all products was performed by comparison with commercial authentic samples or by spectroscopic techniques. For example all 3 compounds show a mass spectrum with a similar fragmentation pattern, with a low abundance molecular ion and a base peak represented by a ion I or II, both of which have greater aromatic character than the molecular ion (Fig. 2).
Scheme 1
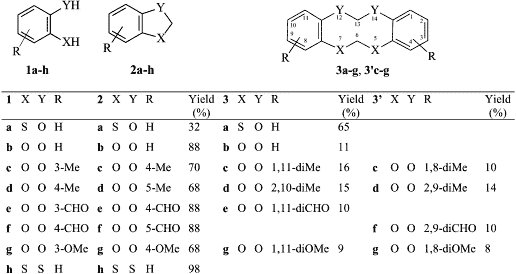
Two isomers 3c and 3'c were identified by 1H NMR analysis of their mixture. The HPLC analysis revealed two peaks with very close retention times (4.38 and 4.59 min, respectively) so that their separation appeared to be very difficult. The 1H-NMR spectrum of their mixture showed two singlets at δ 2.26 and 2.28 in the ratio of 1:1.1, attributable to the methyl groups of the two isomers; two singlets at δ 5.57 and 5.62 were due to the non-equivalents methylenes of the more abundant isomer 3c and another singlet at δ 5.56 was attributed to the two methylenes of the isomer 3’c.
For 3d and 3’d the HPLC analysis of their mixture revealed only one peak. The two isomers were identified by the 1H NMR spectrum of their mixture: two singlets at δ 5.61 and 5.65 were attributed to the two non-equivalents methylenes of the more abundant isomer 3d and a singlet at δ 5.63 was attributed to the two methylenes of 3’d. The relative intensity of these signals revealed a ratio between the two isomers 3d/3’d of 1.1:1. The methyl groups of the two isomers gave only one signal at δ 2.33.
Following the reaction of 1g chromatography gave 2g and an inseparable mixture of 3g and 3’g. Also isolated was a mixture of isomeric dibenzodioxines. The compound 3g was identified by two singlets, due to the methylenes, at d 5.57 and 5.73 in the 1H NMR spectrum, while another singlet at 5.85 was attributed to the equivalent methylenes of 3’g. On the contrary, we were unable to separate the two dibenzodioxines 4 and 5 because their HPLC retention times were too close (5.05 and 5.3 min, respectively): then we performed several successive flash-chromatographies to enrich the mixture in one isomer. We reached a mixture composition of 5:1 and its elemental analysis and mass spectrum were identical to the ones of the 1:1 mixture. The mass spectrum showed the molecular ion as base peak and a very poor fragmentation, typical of aromatic compounds. The enrichment made easier the assignment of some signals in the 1H and 13C NMR spectra, but it was not possible to attribute them to one of the four possible structures. The formation of the benzodioxines was completely unexpected: in fact these compounds are usually obtained through nucleophilic substitution by phenoxide anion on aryl halides or by self-condensation of 2-halogenophenols19,20. We may tentatively justify the formation of these benzocondensed compounds by a radical process involving a semiquinone radical, which can easily be formed in alkaline solution by oxidation. This anion radical then attacks the already formed benzodioxole leading to the condensed 4 (or 4’) and 5 (or 5’).
The molecular ion of 3’f gave a particularly low abundance peak because the two CHO in para to the OCH2O led to a predominance of the symmetric fragmentation showed in the following structure (Fig. 3).
Figure 1
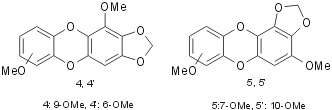
Figure 2-3
Experimental
1.2. Materials
Reagent-grade commercially available reagents and solvents were used. 2-Hydroxybenzenethiol (1a), 1,2-benzodiol derivatives (1b–f) and 1,2-dimercaptobenzene (1g) were purchased (Aldrich).
1.3. General method of methylenation of compounds 1a–h
To a suspension of the starting compound 1a–h (50 mmol), cesium carbonate (24.4 g, 75 mmol) and anhydrous N,N-dimethylformamide (120 mL), was added dropwise bromochloromethane (9.6 g, 75 mmol) and the resulting mixture was vigorously stirred under a nitrogen atmosphere and then heated at 107–110°C for 2 h. The mixture was poured into water, the organic layer separated and the aqueous layer extracted with ether and then with dichloromethane, because ether is able to extract only the monomeric product while the dimeric ones are extracted by dichloromethane. The organic phases were dried (Na2SO4). The solvent was evaporated in vacuo and the residue was analysed by HPLC.
1.3.1. Methylenation of 1a.
The HPLC analysis showed the presence of two products. The extract was flash- chromatographed using light petroleum as eluent. The first fraction was identified as 2a by comparison of its NMR and mass spectra with those of an authentic sample; yield 32%; pale yellow oil, bp 101–102°C/10 mmHg (lit.17 bp 93–95°C/5 mmHg). pale yellow oil, bp 65–66°C/10 mmHg (lit.21 bp 60°C/9 mmHg).
The second fraction was identified as 3b by comparison with an authentic sample; yield 11%; crystallised from acetic acid as white crystals, mp 262°C (lit.21 mp 261–262°C).
1.3.3. Methylenation of 1c.
The HPLC analysis showed three peaks: two peaks had very close retention times (4.38 and 4.59 min, respectively) so that their separation appeared to be very difficult.
The extract was flash-chromatographed using light petroleum as eluent. The first fraction was identified as 2c by comparison of its NMR and mass spectra with those of an authentic sample; yield 70%; pale yellow oil, bp 60°C/3 mmHg (lit.22 bp 45°C/1.2 mmHg). The 1H-NMR analysis of the second fraction showed the presence of two isomers 3c and 3’c in a 1.1:1 ratio; total yield 26%; crystallised from acetic acid as white crystals, mp 190–192°C.
1.3.4. Methylenation of 1d.
The HPLC analysis showed two peaks. The extract was flash-chromatographed using light petroleum as eluent. The first fraction was identified as 2d by comparison of its NMR and mass spectra with those of an authentic sample; yield 68%; pale yellow oil, bp 115–117°C/3 mmHg (lit.21 bp 196–197°C/753.5 mmHg).
The 1H-NMR analysis of the second fraction (corresponding to the second HPLC peak) showed the presence of two isomers 3d and 3’d in a 1.1:1 ratio; total yield 29%; crystallised from acetic acid as white crystals, mp 219–220°C.
1.3.5. Methylenation of 1e.
The HPLC analysis showed the presence of two products. The extract was flash- chromatographed using 10:1 hexane/ethyl acetate as eluent. The first fraction was identified as 2e by comparison of its NMR and mass spectra with those of an authentic sample; yield 88%; crystallised from ethanol as white crystals, mp 32–34°C (lit.23 mp 33°C).
The second fraction was identified as 3e. Yield 10%, crystallised from acetic acid as a white solid, mp 200–202°C.
1.3.6. Methylenation of 1f.
The HPLC analysis showed the presence of two products. The extract was flash- chromatographed using 10:1 hexane/ethyl acetate as eluent. The first fraction was identified as 2f by comparison of its NMR and mass spectra with those of an authentic sample; yield 88%; crystallised from ethanol as white crystals, mp 36–37°C (lit.24 mp 37°C).
The second fraction was identified as 3’f. Yield 10%; crystallised from acetic acid as white crystals, mp 260–262°C.
1.3.7. Methylenation of 1g.
The HPLC analysis showed four peaks. The mixture was flash-chromatographed using 9:1 ether/light petroleum as eluent. The first fraction was identified as 2g by comparison of its NMR and mass spectra with those of an authentic sample; yield 68%; crystallised from ethanol as white crystals, mp 41–42°C (lit.15 mp 39–41°C).
The 1H-NMR analysis of the second fraction (corresponding to the second HPLC peak) shows the presence of two isomers 3g and 3’g in a 1.1:1 ratio; yield 17%; crystallised from methanol as white crystals, mp 205–206°C.
The third fraction was identified as a 1:1 mixture of two products 4 (or 4’) and 5 (or 5’). Yield 10%. This mixture was isolated by chromatography as a white solid with mp 219–220°C.
After repeated column chromatographies on silica using light petroleum as eluent the mixture was enriched in one of the two components reaching a ratio of 5:1. The mp of this mixture was 232–234°C. The elemental analysis and mass spectrum of the 5:1 mixture were identical to those of the 1:1 mixture.
1.3.8. Methylenation of 1h.
The HPLC analysis showed the presence of 2h. The residue was flash-chromatographed using light petroleum as eluent. The first fraction was identified as 2h by comparison of its NMR and mass spectra with those of an authentic sample; yield 98%; pale yellow oil, bp 104–106°C/2 mmHg (lit.25 bp 95–96°C/1 mmHg). We were not able to identify the trace amount products.