2. Experimental
The GC analysis was carried out on a Hewlett Packard 5880A GC equipped with a flame ionization detector. A HP-1 12 m x 0.22 mm I.D. column with a 0.33 µm film thickness was used with He as carrier gas at a pressure of 150 kPa. The injection temperature was held at 250°C and used in the split mode with a split ratio of ~100:1. The detector temperature was 300°C and the following oven temperature program was run: initial temperature 75°C for 1 min, 20°C/min to 300°C, then 300°C for 3 min. Electron impact GC/MS analysis was as reported previously4. Chemical ionisation GC/MS data were obtained on a HP 5890 gas chromatograph interfaced to a HP 5980 mass spectrometer, using methane as the reagent gas.
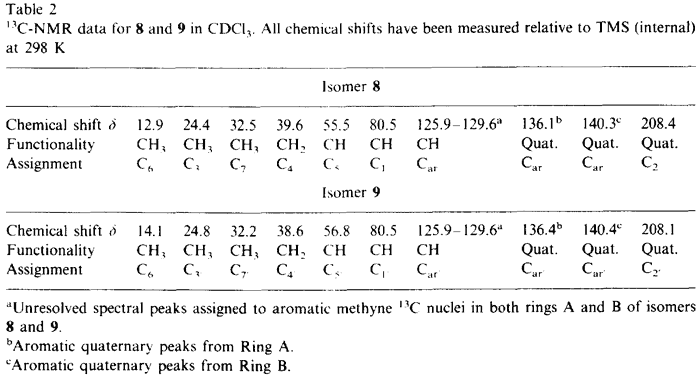
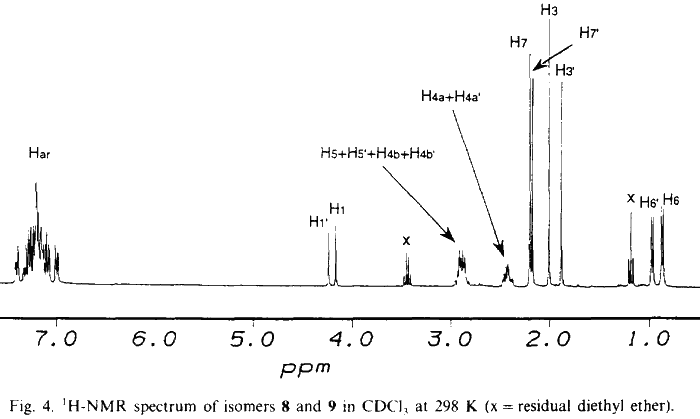
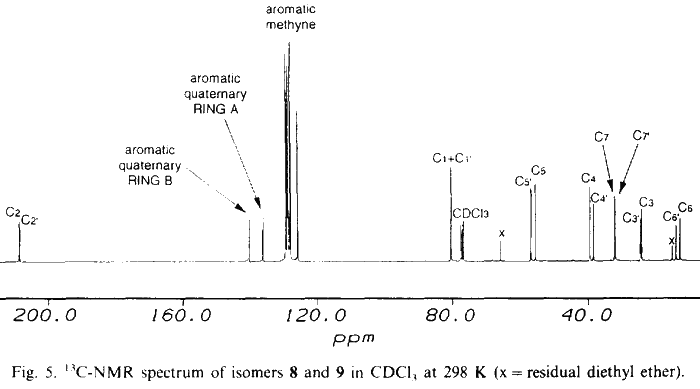
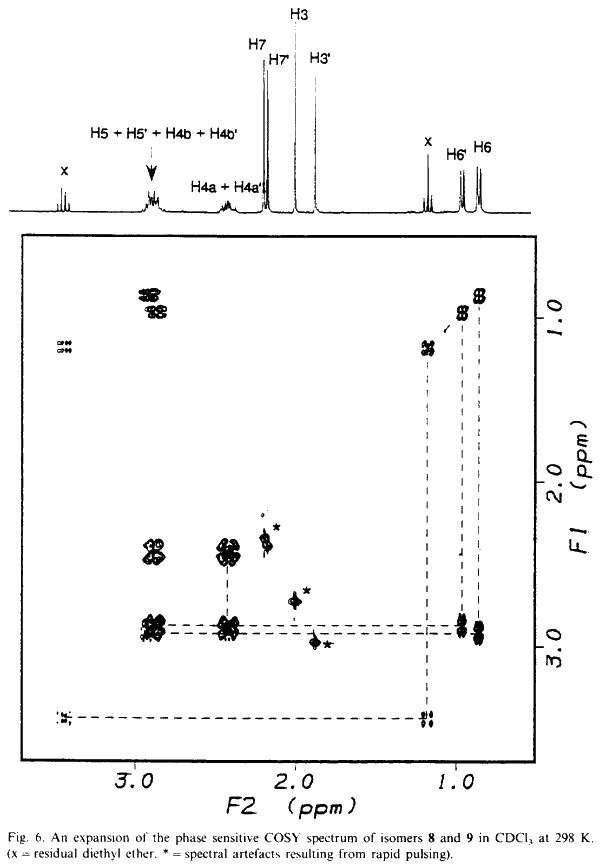
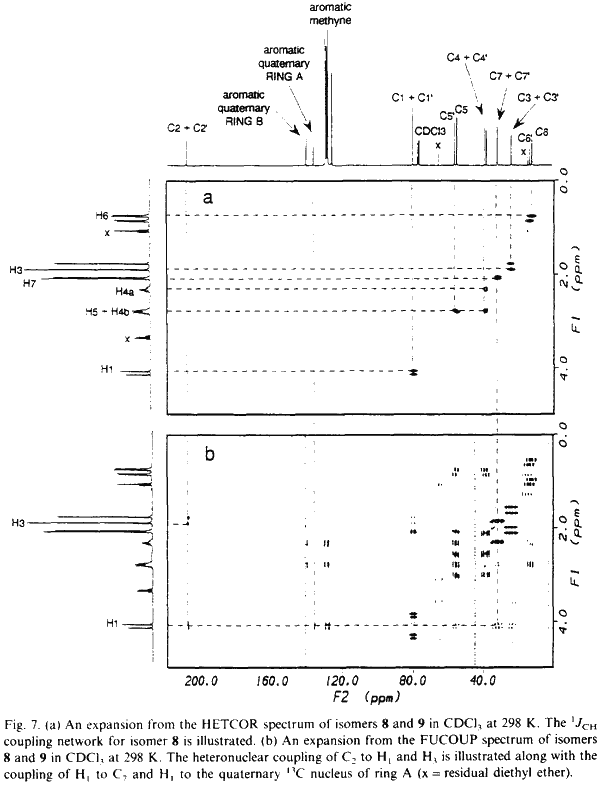
NMR data for 8 and 9 were acquired on a Bruker AM 300 spectrometer equipped with an Aspect 3000 computer, a variable temperature unit and a 5-mm broadband probe at 298 K. 1H-NMR data were acquired at 300.13 MHz over 8K data points with a spectral width of 3400 Hz; 32 transients were collected using a 20° pulse and a recycle delay of 3.3s. 13C-NMR data were acquired at 75.47 MHz over 32K data points with a spectral width of 22700 Hz. Approximately 20000 transients were collected (overnight) using a 30° pulse and a recycle delay of 1.5 s. COSY data were collected using the standard Bruker microprogramme COSYPHDQ.AUR; 2K data points were acquired in F2 with a 1H spectral width of 3400 Hz; 768 experiments of 16 transients were acquired in F1 with a 1H spectral width of 3400 Hz. Data were collected in phase sensitive mode and 90° phase shifted sinebell squared windows were applied in each dimension before transformation. HETCOR data were collected using the standard Bruker microprogramme XHCORRD.AUR; 2K data points were acquired in F2 with a 13C spectral width of 17200 Hz; 554 experiments of 128 transients were acquired in F1 with a 1H spectral width of 3400 Hz. Data were collected in magnitude mode. Line broadening of 3 Hz was applied to F2 and a sinebell squared window was applied to F1 before transformation. FUCOUP data were collected using the pulse sequence as specified by Halterman et al.10; 2K data points were acquired in F2 with a 13C spectral width of 17200 Hz; 768 experiments of 128 transients were acquired in F1 with a 1H spectral width of 3400 Hz. Data were collected in magnitude mode. A 90° phase shifted sinebell squared window was applied to F2 and a sinebell squared window was applied to F1 before transformation.
NMR data for 10 were acquired on a Bruker AM 400 spectrometer equipped with an Aspect 3000 computer, a variable temperature unit and a 5-mm CH probe at 298 K. 1H-NMR data were acquired at 400.13 MHz over 8K data points with a spectral width of 3800 Hz; 32 transients were collected using a 20° pulse and a recycle delay of 3.0 s. 13C-NMR data were acquired at 100.62 MHz over 16K data points with a spectral width of 20000 Hz. Approximately 30000 transients were collected (overnight) using a 20° pulse and a recycle delay of 1.0 s. COSY data were collected using the standard Bruker microprogramme COSY,AUR; 2K data points were acquired in F2 with a 1H spectral width of 3470 Hz; 512 experiments of 16 transients were acquired in F1 with a 1H spectral width of 3470 Hz. Data were collected in magnitude mode; 90° phase shifted sinebell squared windows were applied in each dimension before transformation. HETCOR data were collected using the standard Bruker microprogramme XHCORRD.AUR; 2K data points were acquired in F2 with a 13C spectral width of 20000 Hz; 512 experiments of 256 transients were acquired in F1 with a 1H spectral width of 3470 Hz. Data were collected in magnitude mode. Line broadening of 1 Hz was applied to F2 and a 90° phase shifted sinebell squared window was applied to F1 before transformation. All NMR samples were mixed with deuterochloroform (approximately 10% v/v). The deuterochloroform solvent contained an internal reference of tetramethylsilane (0.03% v/v). Infrared spectra were obtained as KBr discs on a Perkin Elmer 298 Infrared Spectrophotometer.
2.1. Synthesis
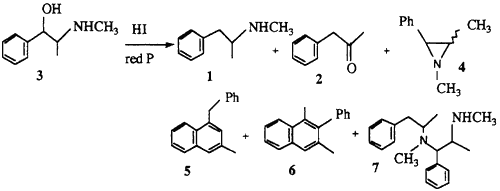
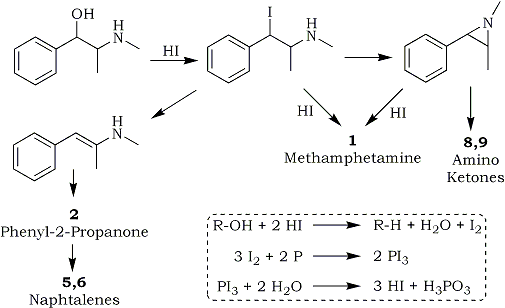
2.1.1. Preparation of methamphetamine 1 and the isolation of impurities
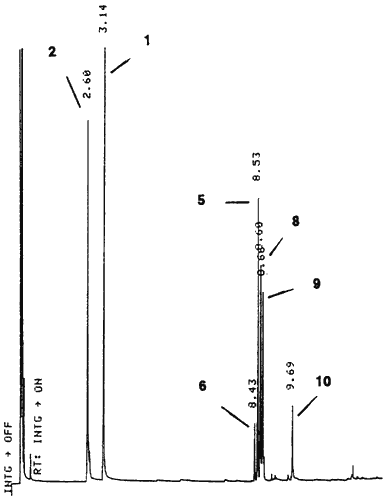
A solution of pseudoephedrine hydrochloride (5 g), red phosphorus (0.1 g) and hydriodic acid (10 ml) was heated under reflux for 5 h followed by the addition of water (12 ml). After standing overnight, the reaction mixture was rendered basic with 20% NaOH and the organic products extracted into ether. The ether extract was filtered and HCl gas was passed through the solution to precipitate the majority of the methamphetamine as the HCl salt. The ether was removed by evaporation to give a residual oil which was analysed by GC (Fig. 2). This oil was redissolved in ether and the basic compounds 8 and 9 and the residual methamphetamine were extracted with 10% HCl. The neutral impurities (mainly 2, 5, 6 and 10) remained in the ether solution. The individual impurities were further separated and purified by chromatography on silica.
2.1.2. Cis- and trans-1,2-dimethyl-3-phenylaziridine 4
Chloropseudoephedrine hydrochloride (2 g), triethylamine (4.5 ml) and acetonitrile (100 ml) were heated under reflux for 30 min and the resulting solution evaporated in vacuo to give the aziridine. Hexane and 5% NaHCO3 were added and the hexane layer separated. Removal of the hexane by evaporation gave a mixture of the cis and trans aziridines (64%) in a ratio 3:1 (cis/trans). The spectral data was in good agreement with the reported data3.
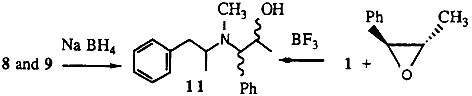
2.1.3. N-methyl-N-(α-methylphenethyl)amino-1-phenyl-2-propanol 11
To trans-2-methyl-3-phenyloxirane (0.5 g) and methamphetamine (1 g) in diethyl ether, boron trifluoride diethyl etherate (0.52 g) was added dropwise with stirring. After stirring for a further 20 min, the reaction mixture was heated under reflux for 20 min. A 10% HCl solution was added followed by enough 20% sodium hydroxide to render the solution basic. The organic products were extracted into ether. Removal of the ether gave the desired alcohols (0.9 g) as an approximately 1:1 mixture of diastereoisomers.
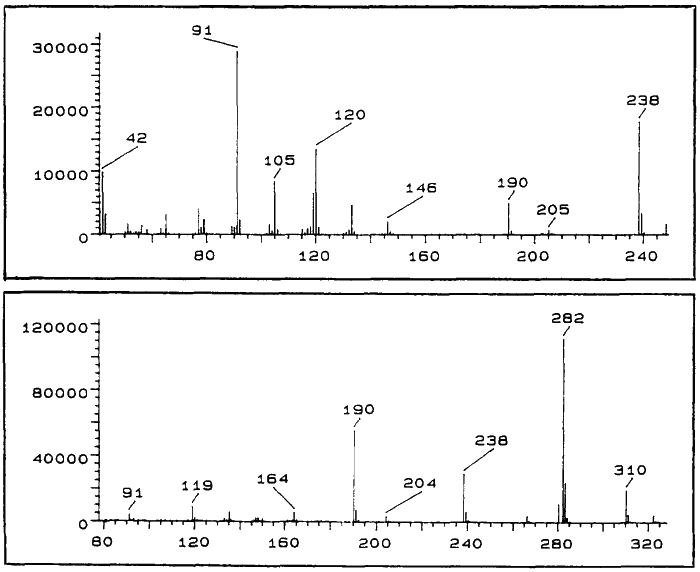
CI-MS: 11A m/z:
284 (100%, M+1), 266 (46%) 238 (39%), 192 (94%), 150 (72%), 135 (37%), 119 (69%), 91 (59%)
CI-MS: 11B m/z:
284 (83%, M+1), 266 (46%), 238 (35%),192 (100%), 150 (48%), 135 (41%), 119 (50%), 91 (37%)
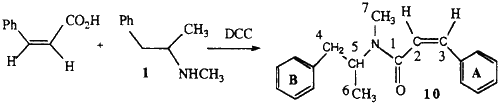
2.1.4. (Z)-N-methyl-N-(α-methylphenethyl)-3-phenylpropenamide 10
Cis-cinnamic acid was prepared by the hydrogenation of 3-phenylpropiolic acid over Pd/CaCO3 in the presence of quinoline. This acid (0.8 g), methamphetamine (0.8 g) and 1,3-dicyclohexylcarbodiimide (1.6 g) in dichloromethane were stirred at room temperature for 30 min. Hexane was added and the dicyclohexylurea removed by filtration. The organic layer was washed with 5% HCl and 5% NaHCO3 to remove any unreacted staining materials and the solvent removed by evaporation. The crude product was purified by chromatography on silica.
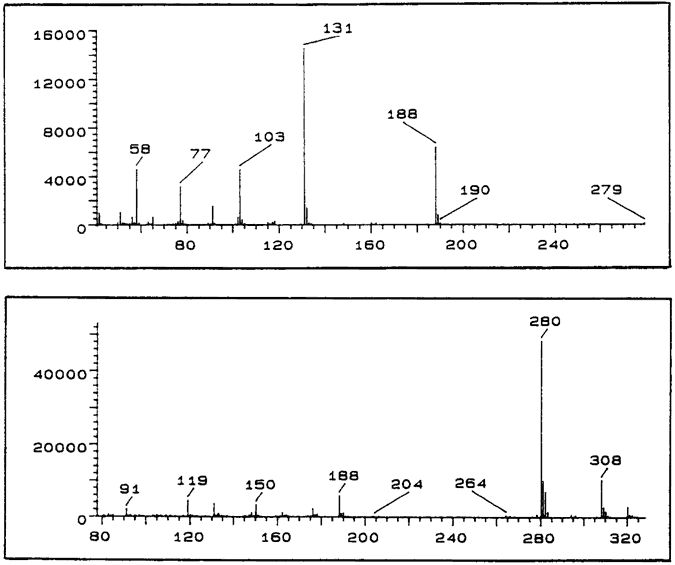
EI-MS m/z: 279 (2%), 188 (56%) 131 (100%), 103 (30%), 91 (10%), 77 (19%), 58 (21%).
2.1.5. (E)-N-methyl-N-(α-methylphenethyl)-3-phenylpropenamide 12
Methamphetamine (1 g) was added to 10% sodium hydroxide solution in a small stoppered flash trans-cinnamoyl chloride (1 ml) was added in small portions, accompanied by vigorous shaking. The mixture was extracted with chloroform and the chloroform evaporated to give the product as an oil.
EI-MS m/z: 279 (2%), 188 (37%), 131 (100%), 103 (34%), 91 (11%), 77 (23%), 58 (20%).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}