Abstract
3,4-Methylenedioxymethylamphetamine (MDMA) was prepared by three synthetic routes. Analytical data from thin-layer chromatography, gas chromatography and gas chromatography-mass spectrometry of the precursors (safrole and isosafrole), intermediates (isosafrole glycol, piperonylmethylketone, N-formyl-3,4-methylenedioxymethylamphetamine, N-formyl-3,4-methylenedioxyamphetamine and 1-(3,4-methylenedioxyphenyl)-2-bromopropane), reaction by-products and the product MDMA were obtained. Further analyses of MDMA using other techniques including 1H- and 13C-nuclear magnetic resonance spectroscopy, X-ray diffraction, infrared spectroscopy, ultraviolet spectroscopy and high performance liquid chromatography were also carried out. The results were then used as reference data for the identification of MDMA in case samples and also to establish the route of synthesis of illicitly prepared MDMA by the study of trace impurities.
Introduction
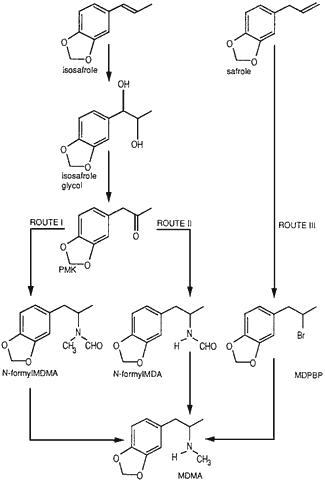
Fig. 1.
Diagram of the synthetic routes investigated.
Route I: PMK → N-formyl-MDMA → MDMA
Route II: PMK → N-formyl-MDA → MDMA
Route III: safrole → MDPBP → MDMA.
Although patented in 1914 as an appetite suppressant1, 1-(3,4-methylenedioxyphenyl)-2-(N-methylamino)- propane, more commonly known as 3,4-methylenedioxymethylamphetamine (MDMA or Ecstasy), is a relatively new drug of abuse in the UK. Over the last 4 years, the number of seizures of the drug has increased and already, illicit laboratories for the production of MDMA have been uncovered. There is no current therapeutic use for MDMA in the UK and under British legislation, it is controlled as a class 'A' drug by the Misuse of Drugs Act, 1971, as amended by the Misuse of Drugs Act, 1971 (Modification) Order, 1977.
MDMA in illicit preparations was first observed in 1972 (Gaston, T.R. and Rasmussen, G.T. pers. commun.) and various aspects of this drug have been reviewed2,3. However, there is only limited information on the forensic examination of the various methods of illicit manufacture. Verweij4 examined the reaction mixtures of MDMA prepared by low pressure reductive amination. A number of impurities were identified by gas chromatography-mass spectrometry (GC-MS). The analyses by GC-MS of samples from a clandestine laboratory involved in the synthesis of MDMA from sassafras oil was carried out by Noggle et al.5. Lukaszeweski6 made a study of the various syntheses of 3,4-methylenedioxyamphetamine (MDA) where the precursors, intermediates and reaction by-products were characterised by chromatographic and spectroscopic techniques.
This study was designed to obtain analytical data pertaining to the identification of precursors, intermediates and reaction by-products encountered in certain synthetic routes as well as obtaining further analytical information to assist in the laboratory identification of MDMA.
Experimental
Materials
Safrole (97%), N-methylformamide (NMF, 99%) and trifluoroacetic anhydride (TFAA) were purchased from Aldrich Chemical Co. (Gillingham, Dorset, UK); isosafrole (cis and trans, 95%) and methylamine (33% in ethanol) were obtained from Fluka Chemical Co. (Glossop, UK); formamide and lithium aluminium hydride (LAH) were from Cambrian Gases (Croydon, UK). Other reagents, solvents (general purpose reagent grade) and methanol (Analar grade) for high-performance liquid chromatography (HPLC) were obtained from BDH Ltd (Poole, UK).
Syntheses
Fig. 1 shows a summary of the syntheses of which Routes I and II involve the Leuckart reaction7,8. The original synthesis of MDMA1 was carried out by Route III. Piperonylmethylketone (PMK) was synthesized from isosafrole through the intermediate isosafrole glycol6.
Route I.
Formic acid (3.66 g), NMF (7.6 g) and PMK (9.0 g) were refluxed at 150ó170∞C for 7 h with additional formic acid (7.32 g) added periodically. On cooling, a clear yellow solution of N-formyl-3,4-methylenedioxymethylamphetamine (N-formyl-MDMA) was obtained. Concentrated hydrochloric acid (30 ml) was added to this solution which was refluxed for a further 3 h. The reaction mixture was made basic with sodium hydroxide and the crude MDMA extracted into diethyl ether. After the volume of the organic solvent was decreased, the remaining residue was treated with hydrogen chloride gas to yield a gelatinous brown precipitate of impure MDMA hydrochloride. The crude salt, dissolved in boiling methanol, was added to chilled acetone to form a crystalline product. This was recrystallized to yield fawn crystals with a melting point of 147ó148∞C9.
Route II.
Formamide (65 g) and PMK (23 g) were refluxed at 190∞C for 5 h. The solution was made basic and extracted with diethyl ether. The ethereal solution was first washed with dilute sulphuric acid, rinsed with water and finally dried over anhydrous sodium sulphate. The diethyl ether volume was reduced to yield a clear yellow solution of N-formyl-3,4-methylenedioxyamphetamine (N-formyl-MDA). This was added drop-wise to LAH (2.5 g in 100 ml of sodium-dried diethyl ether) and refluxed for 3 h. The excess LAH was decomposed by the addition of water and the resulting mixture was filtered and the precipitate washed with diethyl ether. The washings and the filtrate were combined and extracted with dilute sulphuric acid. The aqueous solution was made alkaline with dilute sodium hydroxide and extracted with diethyl ether. The solvent was evaporated leaving an amber oil of crude MDMA.
Route III.
The reactions described in the Merck patent1 involve the formation of 1-(3,4-methylenedioxyphenyl)-2-bromopropane (MDPBP) from safrole followed by reaction with methylamine. This was the method followed.
Extraction of intermediates and reaction by-products
Route I. An aliquot of the acidic N-formyl-MDMA reaction mixture was washed with diethyl ether, made basic with dilute sodium hydroxide and extracted with diethyl ether. This sample was analysed by thin-layer chromatography (TLC), gas chromatography (GC) and GC-MS.
Route II. A sample of the N-formyl-MDA reaction mixture was made acidic with tartaric acid10 and extracted with diethyl ether. The organic layer was separated and extracted with dilute hydrochloric acid. The acidic extract was then made basic with dilute sodium hydroxide and extracted with chloroform. This sample was analysed by the above-mentioned methods.
Route III. A sample of the chloroform used to extract the brominated intermediate from the safrole/hydrobromic acid reaction mixture was analysed by the techniques described above.
Preparation of the trifluoroacetyl (TFA) derivative
Ethyl acetate (1.0 ml) and TFAA (0.1 ml) were added to a 10-ml screw-topped tube containing the dried extract. The reaction mixture was heated at 60∞C for 20 min, evaporated to dryness and methanol (0.1 ml) was added to the tube prior to GC-MS analysis.
Extraction of impurities from case samples
Powder or crushed tablet (5 mg), known to contain the MDMA salt, was vortex-mixed with redistilled diethyl ether (1 ml) and centrifuged. The supernatant was taken off, evaporated to dryness and methanol (0.1 ml) was added prior to GC-MS analysis.
Analytical techniques
HPLC
The two HPLC systems used 12.5 cm by 4.9 mm (i.d.) stainless steel columns with slurry packed 5 µm Spherisorb silica (Phase Separations, Queensferry, UK) and an eluent flow rate of 2 ml/min.
System I. A reciprocating pump, type HM (Metering Pumps, London, UK), delivered the eluent of methanol/30% hydrochloric acid/ammonium hydroxide (d. 0.880) 2000:5.8:18.4. The sample in 0.02 M methanolic hydrochloric acid was introduced to a Rheodyne model 7125 injection valve (Berkeley, CA, USA) fitted with a 5-µl loop. A Cecil CE212 UV detector (Cecil Instruments, Cam-bridge, UK) monitored the eluant by absorption at 284 nm.
System II. An Applied Chromatography Systems model 400 pump (ACS Ltd., Macclesfield, UK) delivered the eluent of 0.01 M ammonium perchlorate in methanol adjusted to pH 6.7 by the addition of 1 ml/l of 0.1 M sodium hydroxide in methanol. The sample in methanol was introduced to a Rheodyne injection valve, model 7125, fitted with a 20-µl loop. A LDC spectromonitor III (LDC Analytical Ltd., Stone, UK) monitored the eluant by UV absorption at 284 nm.
GC-MS
A VG 12-12F quadrupole mass spectrometer (VG Biotech, Altrincham, UK) was used in combination with a Carlo Erba model 4160 gas chromatograph (Fisons Instruments, Crawley, UK). The inlet of a fused-silica capillary column of bonded dimethyl silicone (15 m by 0.22 mm i.d., 0.25 µm film thickness; Thames Chromatography, Maidenhead, UK) was connected to a split/splitless injector. The column outlet was inserted directly into the ion source of the mass spectrometer. A splitless injection was made with the GC oven temperature held at 100∞C for 1 min. The temperature was ramped at 30∞C/min to 280∞C where it was maintained for 5 min. The temperature of the injection port and the transfer line was 270∞C. The inlet pressure for the helium carrier gas was 1.0 kg/cm-2. The mass spectrometer was used in the EI mode, the source temperature was 200∞C and electron energy was 70 eV. Mass spectra were obtained by scanning from 35 to 535 amu at 1 s/scan and the data was processed on a VG DS2050 Data System (VG Analytical, Manchester, UK). Isobutane was the reagent gas in the chemical ionization (CI) mode and the source temperature was 200∞C. The vacuum in the source housing was 10-4 Torr and the mass spectrometer scanned from 100 to 400 amu at 1 s/scan.
Other techniques
Ultraviolet (UV) spectra were recorded on a Uvikon UV/visible spectrophotometer, model 810 (Kontron Scientific Instruments Ltd., St Albans, UK). Infrared (IR) spectra were obtained as potassium bromide discs on a Perkin Elmer IR spectrophotometer, model 298, used in conjunction with a Perkin Elmer IR data station 3600 (Perkin Elmer Instruments, Beaconsfield, UK). The other techniques are detailed with their tabulated data.
Results and Discussion
MDMA and intermediate compounds
There is a large number of potential synthetic routes to MDMA2 but the choice of this study was restricted to three. From the information available, it appears that the Leuckart reaction (Routes I and II) is the most commonly used reaction in the illicit production of amphetamine type drugs. This reaction can be easily adapted to the manufacture of methylenedioxy-substituted analogues. The reactions (Route III) described in the Merck patent1 could be used for those in search of a published method.
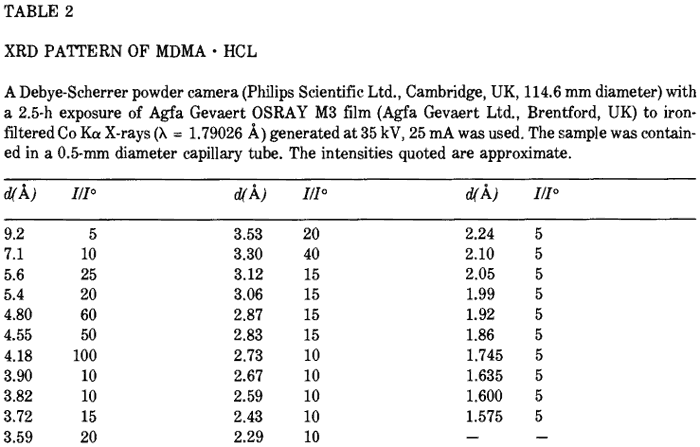
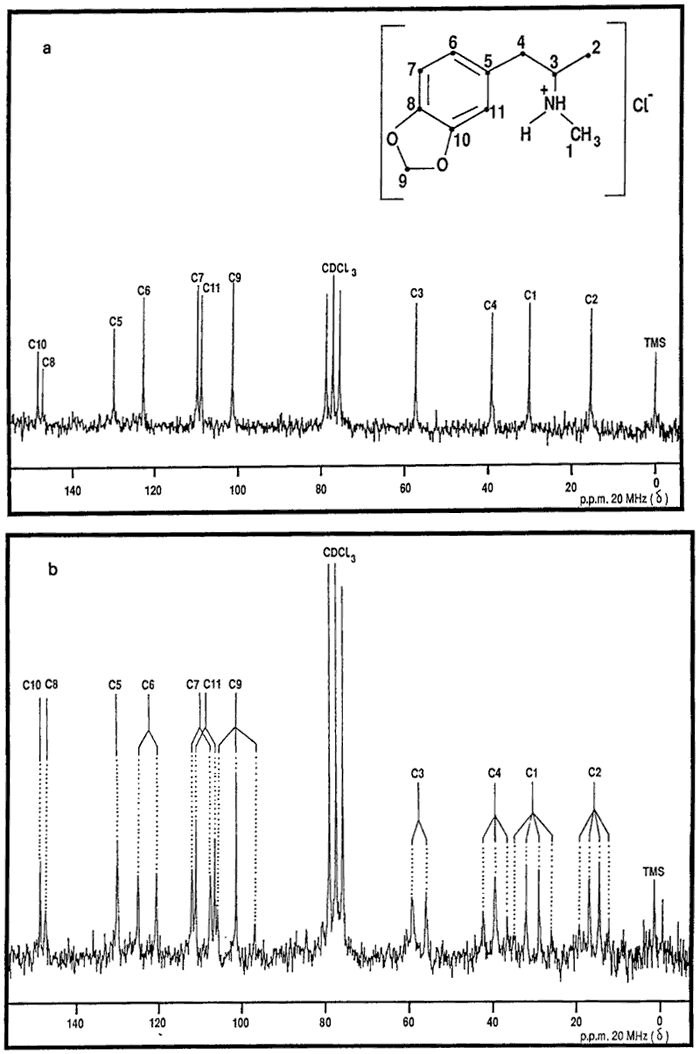
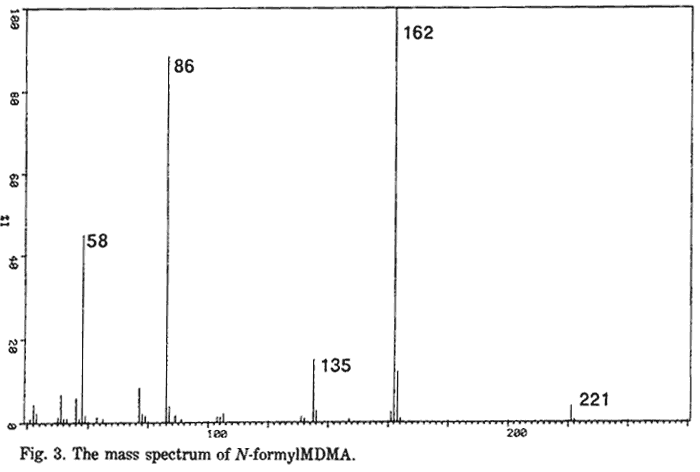
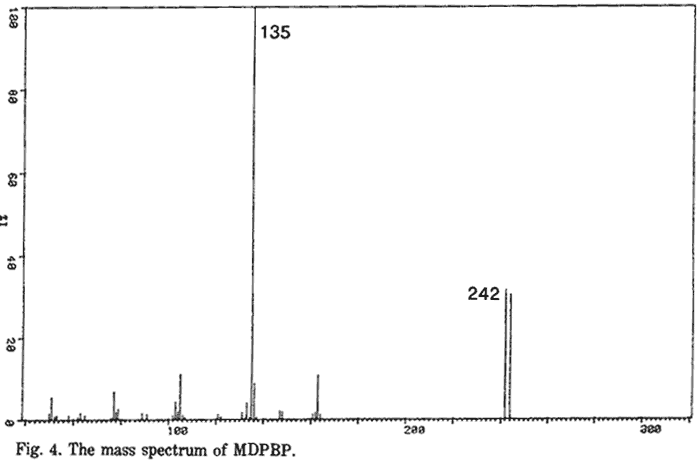
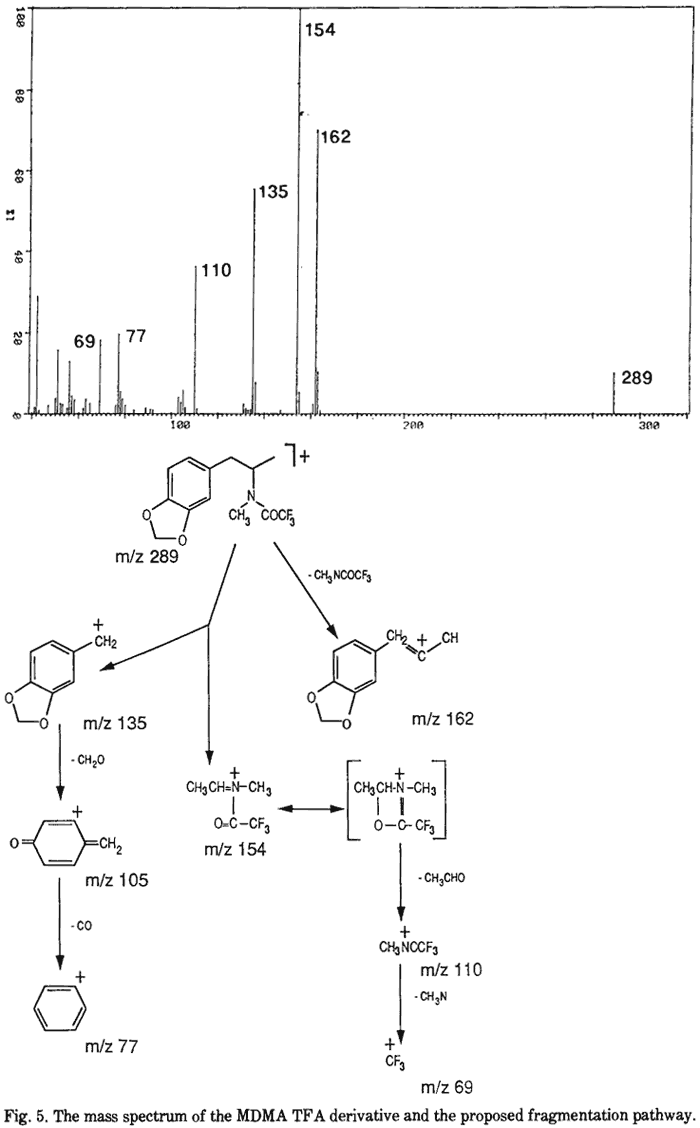
The IR, UV and 1H-MMR spectra were all consistent with those published previously9,13. The 13C-NMR spectra (both broad-band proton decoupled (BBPD) and single frequency off-resonance decoupled (SFORD) spectra) and the XRD pattern of MDMA·HCl are presented in Fig. 2 and Tables 1 and 2 respectively. Fig. 3 and 4 show the mass spectra of N-formyl-MDMA and MDPBP. The mass spectra of safrole, isosafrole, isosafrole glycol, PMK, N-formyl-MDA and MDMA have been reported previously6,9. The mass spectrum of MDMA is not highly characteristic. It has a base peak of mass 58 with minor ions of mass 135 and 13614. A highly characteristic mass spectrum can be obtained with the TFA derivative. Fig. 5 shows the mass spectrum and proposed fragmentation route. The chromatographic data for MDMA and related compounds by TLC, GC and HPLC are shown in Tables 3, 4 and 5, respectively.
Reaction by-products
Route I.
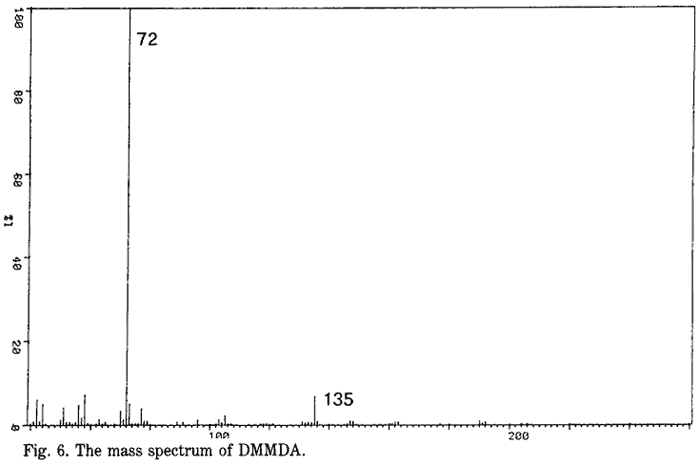
A reaction by-product N,N-dimethyl-3,4-methylenedioxyamphetamine (DMMDA) was identified. This assignation was based on the mass spectrum shown in Fig. 6 together with a molecular weight of 207 (from the CI mass spectrum) and by analogy with the corresponding amphetamine synthesis11,12. However, DMMDA (a tertiary amine) has the same mass spectrum as its isomer N-ethyl-3,4-methylenedioxyamphetamine (a secondary amine)13,14. The compound did not, however, form a derivative with TFAA, showing it to be a tertiary amine and not a secondary amine. This compound could be the product of the reaction of dimethylformamide (DMF) and PMK where DMF is an impurity of NMF12. Verweij4 has identified DMMDA as an impurity in illicit MDMA manufactured by low pressure reductive amination.
Route II.
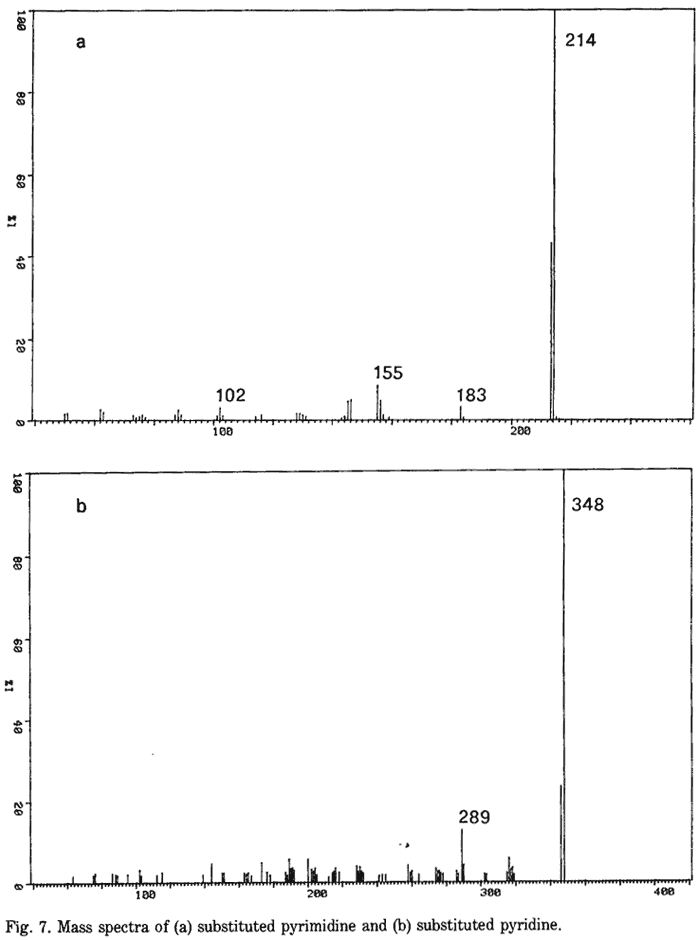
Reaction by-products such as [1-(3,4-methylenedioxyphenyl)-2-propyl]amine and [1-(3,4-methylenedioxyphenyl)-2-propyl]methylamine which have been identified in the synthesis of MDA6 using the Leuckart reaction were not observed in this study. However, GC-MS provides some evidence to suggest the presence of methylenedioxy substituted pyrimidines and pyridines analogous to those observed in the cognate synthesis of amphetamine10,15. The mass spectrum of the tentatively substituted pyrimidine (Fig. 7a) is characterized by its two major ions of mass 213 and 214. The molecular ion of mass 214 exhibits a mass shift of 44 from that observed in the mass spectrum of the cognate amphetamine impurity15, suggesting a methylenedioxy substituted analogue. Similarly, the molecular ion of mass 348 (Fig. 7b) of the tentatively identified substituted pyridine impurity10 exhibits a mass shift of 88 with respect to similar compounds observed in amphetamine synthesis. This suggests a methylenedioxy doubly substituted pyridine analogue which is consistent with the structure of the known impurity.
No significant reaction by-products were identified in Route III which involves the formation of the intermediate MDPBP.
Applications
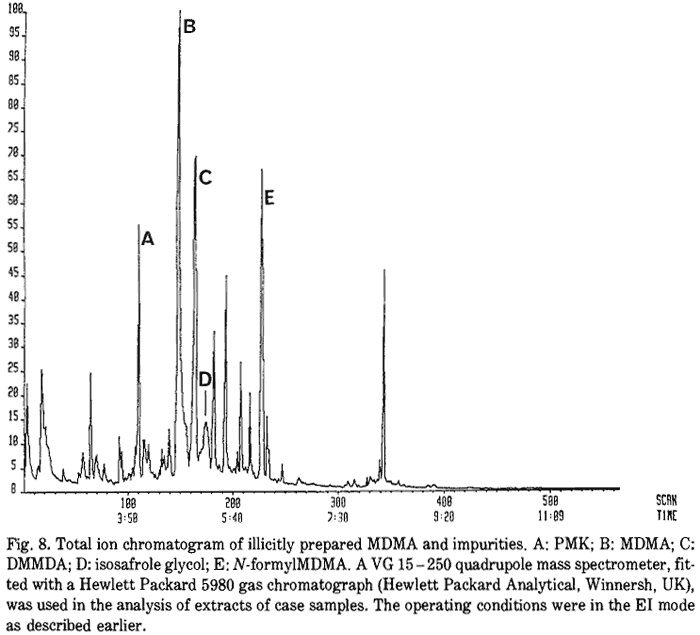
Impurities are often observed in illicitly prepared drugs samples as they are not usually purified to any great degree after manufacture. In the case of illicitly prepared amphetamine, the presence of 'route specific' impurities is used to establish the manufacturing process11. Applying this analogy to MDMA, the identification of the reaction intermediates in the illicit sample can be used to establish the synthetic route used. Figure 8 shows the chromatogram of an extract of a typical sample. The reaction intermediates isosafrole glycol, PMK and N-formyl-MDMA were observed together with the reaction by-product DMMDA. This demonstrates that route I was used in the manufacture.
Conclusion
In addition to providing reference data for the identification of MDMA in case samples, the analytical data from this study can be used to determine the synthetic route used in the illicit manufacture of MDMA, and may also help in the discrimination of sources of origin in the comparison of illicit samples.
Table 4
GC Data for MDMA and Related Compds.
Compound |
Relative retention time |
(MDMA = 1.0) | |
Safrole | 0.35 |
trans-Isosafrole | 0.44 |
cis-Isosafrole | 0.53 |
PMK | 0.97 |
DMMDA | 1.21 |
MDPBP | 1.44 |
Isosafrole glycol | 2.24 |
N-formyl-MDA | 5.77 |
N-formyl-MDMA | 6.45 |
GC was performed using a glass column
(2 m x 6 mm o.d.) packed with 3% OV17
coated on GasChromQ (Phase Sep.,
Queensferry, UK) with a N2 carrier gas
flow rate of 30 ml/min. The Philips
PU4500 gas chromatograph was fitted
with a flame ionization detector. The
oven temp was 200∞C isothermal,
injector temp was 200∞C and the
detector temp was 280∞C.
Table 1
Data From 13C-NMR Spectra of MDMA∑HCl
Chemical shift (BBPD, Fig. 2a) | Multiplicity (SFORD, Fig. 2b) | Assignment | 15.4 | quartet | C-2 |
30.2 | quartet | C-1 |
39.1 | triplet | C-4 |
57.3 | doublet | C-3 |
101.2 | triplet | C-9 |
108.7 | doublet | C-11 |
109.6 | doublet | C-7 |
122.6 | doublet | C-6 |
129.8 | singlet | C-5 |
146.9 | singlet | C-8 |
148.1 | singlet | C-10 |
Table 3
TLC Data for MDMA and Related Compounds
Compound |
Relative Retention Value |
MDMA | 0.27 |
DMMDA | 0.38 |
N-formyl-MDA | 0.75 |
N-formyl-MDMA | 0.78 |
TLC was carried out on glass plates coated
with Kieselguhr 60 F254 (Merck TLC plates,
BDH Ltd., Poole, UK) that had been dipped
in a methanolic solution of 0.01 M sodium
hydroxide16. The plates were developed
in a methanol/acetone (3:1) solvent system,
visualised in ultraviolet light (254 nm) and
sprayed with acidified iodoplatinate solution.
Table 2
![]()
XRD Pattern of MDMA∑HCl
{kind=link}
- Fig. 2. The 13C NMR spectra of MDMA.HCl. (a) BBPD and (b) SFORD.
- Fig. 3. The mass spectrum of N-formyl-MDMA.
- Fig. 4. The mass spectrum of MDPBP.
- Fig. 5. The mass spectrum of the MDMA·TFA derivative and the proposed fragmentation pathway.
- Fig. 6. The mass spectrum of DMMDA.
- Fig. 7. Mass spectra of (a) substituted pyrimidine and (b) substituted pyridine.
- Fig. 8. Total ion chromatogram of illicitly prepared MDMA and impurities. A: PMK; B: MDMA; C: DMMDA; D: isosafrole glycol; E: N-formyl-MDMA. A VG 15ó250 quadrupole mass spectrometer, fitted with a Hewlett Packard 5980 gas chromatograph (Hewlett Packard Analytical, Winnersh, UK), was used in the analysis of extracts of case samples. The operating conditions were in the EI mode as described earlier.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}